Focal segmental glomerular sclerosis (FSGS) is the most common form of primary glomerular scarring. It typically involves segments of scattered glomeruli throughout the kidney and only few glomeruli are affected earlier during the course of the disease process.
It may arise due to a systemic condition or may be idiopathic, which does not have an identifiable cause. Asymptomatic proteinuria or nephrotic syndrome are the clinical features of FSGS. It is a progressive form of renal disease and accounts for as many as 2.3% of all cases of end-stage renal disease (ESRD).
There is a higher prevalence of FSGS among African Americans and males tend to be affected more often in comparison to females. Many patients are asymptomatic, but some may notice swelling of the legs and ankles (i.e., edema) and have hypertension.
Renal biopsies are necessary to confirm diagnosis and treatment may be done using steroids and immune suppressive drugs. However, it is very difficult to prevent the eventual progression to total renal failure, which typically occurs within 5 to 20 years in many cases or in 3 years with more aggressive forms of the disease.
Etiological classification and causes
According to etiology, FSGS can be classified into primary and secondary FSGS. They may be distinguished based on histological and clinical grounds. Urinary excretion of protein within the nephrotic range that is not accompanied by full blown nephrotic syndrome is suggestive of FSGS of a secondary cause.
Secondary FSGS is associated with gradually increasing amounts of protein in the urine, which is initially below nephrotic levels. In secondary FSGS, there is also higher serum levels of albumin and lower serum cholesterol as well as lower edema rates.
It is imperative to make a distinction between primary and secondary FSGS for therapeutic purposes. Immunomodulatory and intraglomerular hypertension reduction are used for primary and secondary FSGS, respectively.
Primary FSGS is mediated by undefined factors that cause dedifferentiation and injury of the podocytes. These are cells in the Bowman’s capsule, which are responsible for the filtration of small molecules like salts and the retention of larger molecules like proteins.
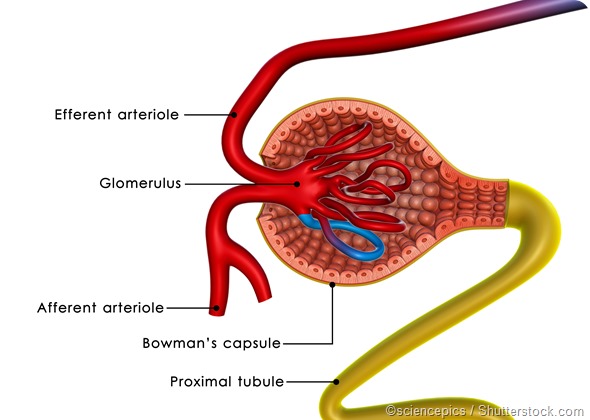
Damage to the filtration mechanism in FSGS leads to abnormal glomerular permeability and the leakage of proteins into the urine. Approximately 8 in every 10 cases of FSGS are due to a primary or idiopathic cause.
Secondary FSGS can be due to many causes, mainly familial/ genetic, viral, drug-induced and functional-structural adaptive responses. Mutations in genes that encode proteins, such as podocin and nephrin have been implicated as secondary causes of FSGS. Viruses such as parvo B1, EBV, CMV, HIV, Simian virus 40, and hepatitis C have been linked to FSGS. Heroin, lithium, and anabolic steroids are also implicated as drug-induced causes.
Conditions such as low birth weigh, obesity, malignant hypertension, renal artery stenosis, and chronic allograft nephropathy, among others, mediate an adaptive functional-structural response and are postulated causes of secondary FSGS.
Histological classification
Histologically, according to the Columbia classification, there are 5 variants of FSGS: classical also known as FSGS not otherwise specified (FSGS NOS), collapsing, tip, perihilar and cellular variants.
FSGS NOS patients present with proteinuria below nephrotic levels. Those with the collapsing or tip variants tend to be older and have higher levels of proteins in the urine.
The tip and collapsing variants may be distinguished from each other by the latter having a higher progression rate and elevated levels of baseline creatinine. In addition, the tip variant is associated with Caucasian, whereas the collapsing variant with African Americans.
The perihilar form of FSGS is seen with secondary adaptive functional-structural responses, while the cellular variant is seen in both primary and secondary FSGS.
Further Reading
Last Updated: Dec 30, 2022