Spinal muscular atrophy (SMA) is a genetic disorder caused by a homozygous recessive mutation in a gene on chromosome 5 (5q13), called the survival motor neuron 1 (SMN1) gene. It is due to deletion (in 95% of cases) or gene conversion (SMN1 to SMN2) by nucleotide substitution at one point.
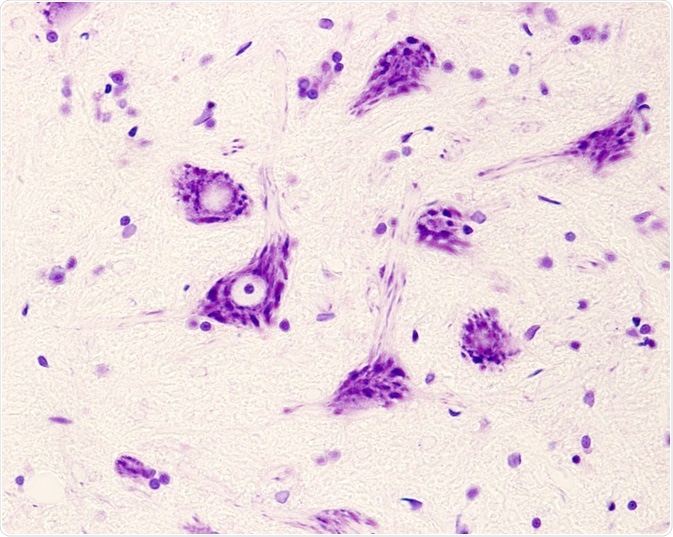
Motor neurons of the anterior horn of the spinal cord. Credit: Jose Luis Calvo/ Shutterstock.com
This absence of the SMN1 gene causes the degeneration of anterior horn spinal motor neuron cells and brain neurons, which results in proximal muscle atrophy and limb weakness.
In over half of cases it has a severe phenotype, but other clinical presentations also occur. The differentiation is made on the basis of onset and the maximum motor function achieved by the patient. A carrier state is also very common, at 1 in 54, while the incidence is 1 in 11 000.
SMN1 and SMN2 genes
It is also now recognized that the various manifestations of SMA are part of a common spectrum of disease with a single etiology. The SMN gene has two forms on each allele, one being telocentric and the other centromeric. These are called SMN1 and SMN2 respectively. The full-length messenger RNA (mRNA) is transcribed from the SMN1 gene, and this gives rise to the SMN protein on translation.
This protein is crucial for the survival of lower motor neurons. The absence of the SMN1 gene on both copies of chromosome 5 gives rise to SMA. These patients must then rely on the SMN2 gene to produce the SMN protein. About 3% of SMA patients have only one SMN1 deletion but the remaining allele shows subtle mutations.
On the other hand, the SMN2 gene is substituted in a single nucleotide (840C > T), which happens to be an exonic splicing enhancer. The result is that exon 7 is not transcribed, which means the production of a truncated protein incapable of function and subject to rapid breakdown.
A small fraction of the SMN2 transcripts, about 10-15%, do have exon 7, however, and so the more copies there are of SMN2, the more chance there is of encoding the normal protein, though in an inefficient manner. Thus, the number of copies of this gene partly decides disease severity.
All patients with SMA have one copy at least of SMN2 and most have 2-4 copies. Type I SMA has two SMN2 gene copies in most cases, while type II patients commonly have 3 copies, and there are 3-4 copies in the least severe forms, type III and IV.
There are some other factors too which modify disease severity, such as other exon 7 mutations which restored the exon splice enhancing element so that more normal SMN protein can be produced, resulting in milder disease.
What is the SMN protein?
SMN is a protein which forms part of an 8-member complex called the SMN complex, present in all cells, and especially rich in the spinal motor neurons. It forms small round structures which are associated with coiled Cajal bodies, and are called gems or gemini of Cajal bodies.
This protein is vital for the formation of a small uridine-rich nuclear ribonuclear protein (U snRNP) which are primary in the formation of the splicesome (an intracellular body which is responsible for pre-mRNA splicing). It is also present in motor neuron axons.
Though it is found in all cells, lowering of its concentration causes fewer gems to be present, or results in a widespread disturbance of snRNP assembly. This seemingly affects motor neuron development and survival selectively, perhaps because the motor neurons produce a unique set of transcribed mRNA that responds differently to this global splicing aberration.
It is also possible that the SMN protein specifically regulates motor neuron function in a unique way (such as transporting RNP complexes or certain mRNAs), and its low concentrations affect these functions deeply. Again, its deficiency may reduce the number and activity of motor neuron synapses, leading to motor neuron loss.
Reviewed by Afsaneh Khetrapal Bsc (Hons)
Further Reading
Last Updated: Aug 23, 2018