A clade, derived from ancient Greek, is a group of organisms that are derived from a common ancestor and its lineal descendants. A clade is also subtypes, genotypes, or groups that all arise from a common ancestor. These relationships can be tracked on her phylogenetic tree.
In virology, viruses are placed in clades based on phylogenetic trees constructed from their genome sequences. These viral clades that share similar genetic sequences and the changes in the viral genome can be tracked mapped using phylogeny.
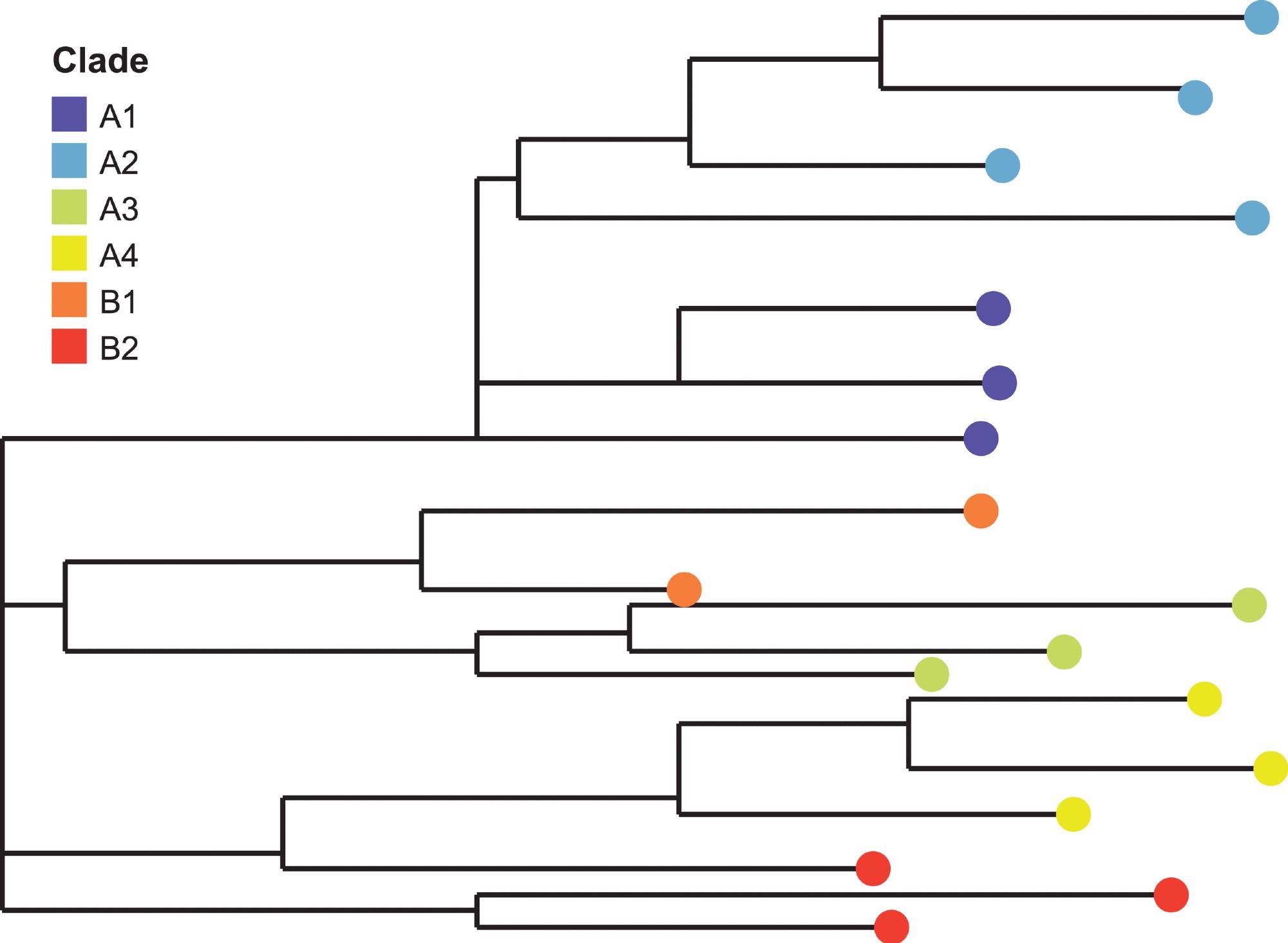
Image Credit: M. PATTHAWEE/Shutterstock.com
The utility of clades and sub-clades and the cladogram
Clades and sub-clades are analogous to ‘groups’ and ‘sub-groups’ respectively. In the case of influenza, which is divided into subtypes based on the two surface proteins: hemagglutinin (HA) and neuraminidase (N). There are 18 different hemagglutinin subtypes and 11 different neuraminidase subtypes.
In the case of the influenza virus, a clade is a further subdivision of influenza viruses based on the similarity of their HA gene sequences. In phylogenetic trees, clades and subclades are viewed as groups of viruses that usually have similar genetic changes. The sorting of viruses into clades and subclades enables the proportion of viruses from different clades in circulation to be tracked.
The cladogram (a visual representation of the phylogenetic relationships) has the oldest (most basal) common ancestor found in a clade positioned close to the base/ trunk of the evolutionary tree. Newly evolved species form the tree branches farthest from the tree trunk. The magnitude of the genetic difference between viruses is proportional to the length of the branches in the phylogenetic tree.
Although clades and subclades are genetically distinct, they may not be antigenically distinct (that is, containing altered proteins or carbohydrates on their surface). Viruses whose genetic sequence share the same genetic changes and a common ancestor are grouped into “clades” and “subclades.” A node demonstrates a common ancestor.
A notable example of a viral clade: SARS-CoV-2
The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is the most recent example of a viral clade. SARS-CoV-2 is clustered with the SARS-CoVs of batS in a clade and is considered to be a virus typical of SARS.
SARS-CoV-2 is a clade within the Coronaviridae family and the betacoronavirus genus. The SARS-CoV-2 genome has been altered as a result of several mutations; of them, 11 major mutations have occurred and are defined in five major clades according to their respective amino acid mutations: D392, S84, L378, V251, and G614.
The SARS-CoV-2 variant carrying the Spike protein amino acid change D614G has become the most prevalent across, the world with the G614 clade being part of the G-clade, which is widespread in several regions and continents (Oceania, Europe, Africa, and South America). Overall, the G clade and its descendants GH and GR, account for 74% of the sequenced SARS-CoV-2 genome.
The clade L was the first SARS-CoV-2 type, appearing in Wuhan, China I 2019. The S clade followed thereafter, which mutated to produce the V clade, surfacing in January 2020, as did the G clade. Nearing the end of February 2020, GR and GH clades appeared and spread globally.
Each clade has distinct characteristics and has been given nomenclature-based names (depending on the system used). The following table outlines the Global Initiative on Sharing All Influenza Data (GISAID) clade characteristics. The GISAID houses all the available genomic sequences for SARS-CoV-2, with over 4,383,873 million SARS-CoV-2 genome sequences (as of 11th November 2021)
|
GISAID Clade
|
Effect on protein
|
|
L
|
nsp 4
|
|
S
|
ORF8 protein
|
|
V
|
nsp 6 (transmembrane protein) and ORF3a protein
|
|
G
|
Spike protein, nsp12, post-ribosomal frameshift (RNA-dependent RNA polymerase), nsp 3 (predicted phosphoesterase)M 5’ Untranslated Region
|
|
GH
|
ORF3a protein
|
|
GR
|
Nucleocapsid protein
|
Since the COVID-19 pandemic, research has been conducted to determine the most ‘basal’ SARS-CoV-2 clade. A basal clade is one closest to the root common ancestor and is the earliest clade to branch in a larger clade. This clade appears at the bottom of a cladogram.
Image Credit: Corona Borealis Studio/Shutterstock.com
Determining the basal clade of SARS-CoV-2
To determine the most basal clade, researchers have identified the mutation sites in the SARS-CoV-2 whole-genome sequence alignment and conducted pair-wise comparisons of the mutation sites among all SARS-CoV-s. From the first analysis of 168 SARS-CoV-2 sequences (GISAID dataset till 2020/03/04), researchers identified a basal clade that contained 33 identical viral sequences across 7 countries. A second analysis (GISAID dataset till 2020/03/17), using 367 sequences from a data set derived a month later than the first analysis, identified a basal clade with 51 viral sequences from 10 countries. This clade had been expanded with 85 unique sequences.
As the earliest collection date for basal SARS-CoV-2 is 2019/12/26 and the latest was 2020/04/04 (in this study), this revealed that the least mutated SARS-CoV-2 sequence has been replicating and spreading for at least 3 months before identification.
These results suggest that the SARS-CoV-2 RNA proofreading capability of the virus is unprecedented, allowing it to preserve its genome even after a long period of transmission.
References:
- Shen S, Zhang Z, He F. (2021) The phylogenetic relationship within SARS-CoV-2s: An expanding basal clade. Mol Phylogenet Evol. doi:10.1016/j.ympev.2020.
- Centers for Disease Control and Prevention (CDC). Types of Influenza Viruses. Available at: https://www.cdc.gov/flu/about/viruses/types.htm. Last accessed November 2021
- Murugan C, Ramamoorthy S, Guruprasad K, et al. (2021) COVID-19: A review of newly formed viral clades, pathophysiology, therapeutic strategies, and current vaccination tasks. Int J Biol Macromol. doi:10.1016/j.ijbiomac.2021.10.144.
Further Reading
Last Updated: Jan 11, 2022
 How pandemic viruses spread across U.S. cities before anyone notices
How pandemic viruses spread across U.S. cities before anyone notices