Barcoding technologies using the high information content encoded in oligonucleotide sequences have aided various applications in biotechnology, especially when paired with next-generation DNA sequencing or NGS, whereby they can decode this information in a low-cost and high-throughput manner.
The combination of DNA barcoding and NGS readout can provide single-cell or spatial resolution in transcriptomics, track sample identity in multiplexed libraries, and monitor the enrichment of genotypes using evolution methods for protein engineering.1–3
Peptide barcodes (PBs) present pioneering opportunities with their easy genetic encoding, ability to encode comprehensive information in short sequences, and chemical versatility. Applications of PBs that utilize mass spectrometry for decoding have been developed, for example, nanobody screening based on flycodes.4
There is a necessity for accessible techniques that can directly read PB sequences and classify single-molecule PBs. Next-Generation Protein Sequencing™ (NGPS) on Quantum-Si's Platinum® instrument enables the novel direct sequencing of single-molecule resolution PBs.
This platform is user-friendly and simple to use, combining DNA-based approaches with the innovation of peptide-based barcoding methods to enable real-time single-molecule sequencing.
The platform’s workflow starts by attaching peptides to macromolecular linkers at the C-terminus for immobilization on a semiconductor chip. After immobilization, dye-labeled N-terminal amino acid (NAA) recognizers and aminopeptidases are introduced to commence sequencing.
These recognizers repeatedly bind and unbind the immobilized peptides when their cognate NAAs are exposed at the N-terminus. This produces a distinct series of pulses for each recognized NAA with distinctive fluorescence and kinetic properties, known as a recognition segment (RS).
The aminopeptidases in the solution sequentially remove individual NAAs, exposing subsequent residues for detection. This process repeats until the peptide has been sequenced to completion.
The temporal order of NAA recognition and associated kinetic properties throughout sequencing are highly characteristic of any given peptide and are called its kinetic signature (KS). KSs are analyzed with Platinum to deliver high-assurance alignments to individual sequences.5
An innovative protein barcoding technique utilizing NGPS on Platinum is presented. A set of synthetic peptides is designed and sequenced, demonstrating that they can be utilized as distinguishable barcodes. The statistical analysis of these sequences enabled the establishment of criteria for PB design and scalability estimation.
A method for enzymatic library preparation of recombinantly expressed PBs is developed, demonstrating the relative quantitation of a variety of PBs on Platinum. The use of PBs to select for proteins with characteristic properties is also demonstrated.
A 300-fold enrichment in the relative abundance of an anti-GFP nanobody (following positive selection using PBs enzymatically cleaved from the enriched nanobody) can be observed.
Methods
Production of synthetic peptides
Using solid-phase peptide synthesis, PBs were synthesized by Innopep. The peptides were primed with an azido-lysine modification at the C-terminus. Using high-performance liquid chromatography (HPLC) and mass spectrometry analysis, peptides were confirmed to be 95% pure.
Barcode expression and purification
PBs were cloned into a plasmid containing an N-terminal HaloTag, a SUMO Tag, a TEV protease site, a Sortase A recognition motif (LPETGG), the PB sequence, and a C-terminal 6x-Histidine tag.
After cloning, the barcode plasmids were transformed into SHuffle T7 express competent E. coli (New England Biolabs,) and cultivated overnight on suitable antibiotic selection media. Using Sanger sequencing, the sequence of the barcode plasmids was confirmed.
Next, 10 mL of Terrific Broth was inoculated with bacteria overnight through shaking at 37 °C. Barcode expression was prompted with the addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) at OD 0.6-0.8. The bacteria were harvested by centrifugation, resulting in a pellet which was washed with 50 mM HEPES pH 7.3 and 150 mM NaCl and stored at –80 °C until purification.
The thawed cell pellets were resuspended in 0.2 mL of 50 mM HEPES, pH 7.3 and 150 mM NaCl. Cells were lysed with the addition of 1 mL of NEB Express lysis buffer and mixed for 30 minutes at room temperature. The bacterial lysate was then centrifuged at 10,000 x g for 10 minutes to pellet cell debris, and the supernatant was collected for purification.
Following this, 200 µL of Ni-NTA resin was pre-equilibrated by washing three times with 50 mM HEPES, pH 7.3, 150 mM NaCl, and 10 mM imidazole buffer. The bacterial supernatant was diluted with 1 volume of 50 mM HEPES, pH 7.3, 150 mM NaCl and 10 mM imidazole buffer.
The supernatant solution was applied to the pre-equilibrated Ni-NTA resin then incubated for 30 minutes at an ambient temperature and mixed. The resin was then pelleted by centrifugation at 1000 x g for two minutes and the supernatant was collected.
The resin was washed three times with 400 µL of 50 mM HEPES, pH 7.3, 150 mM NaCl and 10 mM imidazole buffer. Elution was implemented by adding 500 µL 50 mM HEPES, pH 7.3, 150 mM NaCl, and 300 mM imidazole buffer to the resin, followed by five minutes of incubation at room temperature. The resin was then pelleted by centrifugation at 1000 x g for two minutes.
Sortase A reaction
For the Sortase labeling reaction, 10 µg of purified PB was introduced into a solution containing 1 µM of Sortase A pentamutant (BPS Biosciences), 1 mM of a synthetic tri-glycineazide (GGG-azide) peptide (Click Chemistry Tools) and 1 Sortase Buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl and 10 mM CaCl2).
A final reaction volume of 50 µL was incubated for one hour at 37 °C to facilitate the incorporation of the GGG-azide motif at the C-terminus of the PB.
For the purification of the azide-labeled PB product, 20 µL of Magne HaloTag beads (Promega) were used per barcode. First, the HaloTag beads were washed four times for five minutes with 200 µL of HEB Buffer (HEPES 50 mM pH 7.3, 0.005 % IGEPAL CA-630 and NaCl 150 mM) using a magnetic stand for the bead collection during the wash phase.
Next, 50 µL of HEB buffer was introduced to the 50 µL Sortase reaction at a ratio of 1:1; the combined solution was added to the beads. This mixture was incubated with end-over-end mixing for 1 hour at an ambient temperature.
Following incubation, the beads were separated magnetically, and the flow-through was collected for SDS-PAGE analysis. The beads were washed three times for five minutes with end-over-end mixing using an HEB buffer. Finally, the beads were stored in the final wash buffer until the click chemistry reaction.
Click chemistry reaction
The click reaction was performed while the azide-labeled PBs were still attached to the beads. Following the removal of supernatant solution, 22 µL of HEB buffer was added, and 0.5 µL of Cetyltrimethylammonium bromide (CTAB) and 1 µL of K-Linker from the Library Preparation Kit – Lys-C (Quantum-Si) were introduced. The reaction continued overnight at 37 °C in a Thermomixer (Eppendorf) at 1400 rpm.
The following day, the beads were washed with HEB buffer three times as described. To elute the PBs from the HaloTag, 25 µL of HEB Buffer supplemented with 1 mM DTT and 1 U of Sumo Protease (ThermoFisher) was introduced to each barcode.
The reaction was incubated at 37 °C at 1400 rpm in a Thermomixer for one hour. The beads were then collected and the supernatant containing the eluted PB conjugated to the K-Linker was retrieved. The concentration of the eluted PBs was established using UV-Vis absorbance and gel densitometry.
Selection of nanobodies
The nanobodies targeting GFP (LaG-16) and MBP (Sb_MBP#1) have been characterized.6,7 These constructs were inserted between the SUMO Tag and the TEV protease site in the peptide barcoding plasmid described above.
The MBP nanobody was bonded to Barcode A (RLIFAA) and the GFP nanobody was attached to Barcode B (FLRAA). Next, the barcoded nanobodies were expressed at 22 °C in E. coli, then purified, functionalized with Sortase A, and conjugated via GGG-azide moiety with the macromolecular K-Linker for peptide immobilization on the chip after proteolysis. A pre-selection sample fraction of this library was stored for sequencing using Platinum.
Affinity purification selection was performed on the barcoded model nanobody library using commercially purchased GFP (Sigma-Aldrich) immobilized on M-280 tosylactivated Dynabeads (ThermoFisher). The barcoded nanobody library (10 nM) was incubated with the GFP-coated Dynabeads for five minutes at room temperature in 50 mM Tris pH 7.5 and 0.5 % Tween-20.
Subsequently, the beads were separated magnetically and washed with buffer five times. Barcodes from the post-selection sample associated with the nanobodies that remained on the beads after washing were then harvested by proteolysis, resulting in a nanomolar barcode solution ready for NGPS.
Using platinum for NGPS
Following the user protocols, single-molecule sequencing of PBs was performed: conjugated peptide libraries were loaded onto Quantum-Si’s semiconductor chip through incubation for 15 minutes. A solution containing dye-labeled recognizers was prepared using a sequencing kit (Quantum-Si) and added to the chip.
The chip was then installed in the Platinum instrument and data collection proceeded for 15 minutes. A solution including aminopeptidases from the sequencing kit was added to the chip and mixed; data was collected for 10 hours.
After the sequencing runs were completed, the data was analyzed using the automated Platinum system.
Results and discussion
Short peptide sequences for barcodes on platinum allow the creation of diverse barcode sets
The evaluation of previously sequenced proteins allowed the following peptide sequences to be identified as candidates for barcodes: VRLFEQQN, EFLNRFY, DQRFLAGG, FQIRIALNFA, ARLAFAYDDD, FAQLQARFAADDD, RLAIQFAYPDD and ENRLCYYLGAT.
To examine if this group of peptides had the particular qualities needed for use as a barcode set, synthetic peptides were generated and their sequencing performance was individually analyzed.
Figure 1 shows KS plots for the eight peptides and unique patterns of RSs and on-chip pulse durations. The color in each box refers to the percentage of coverage of observed amino acids in each sequence with the value shown under each box. The figure shows the mean pulse duration for each residue under each peptide sequence box.
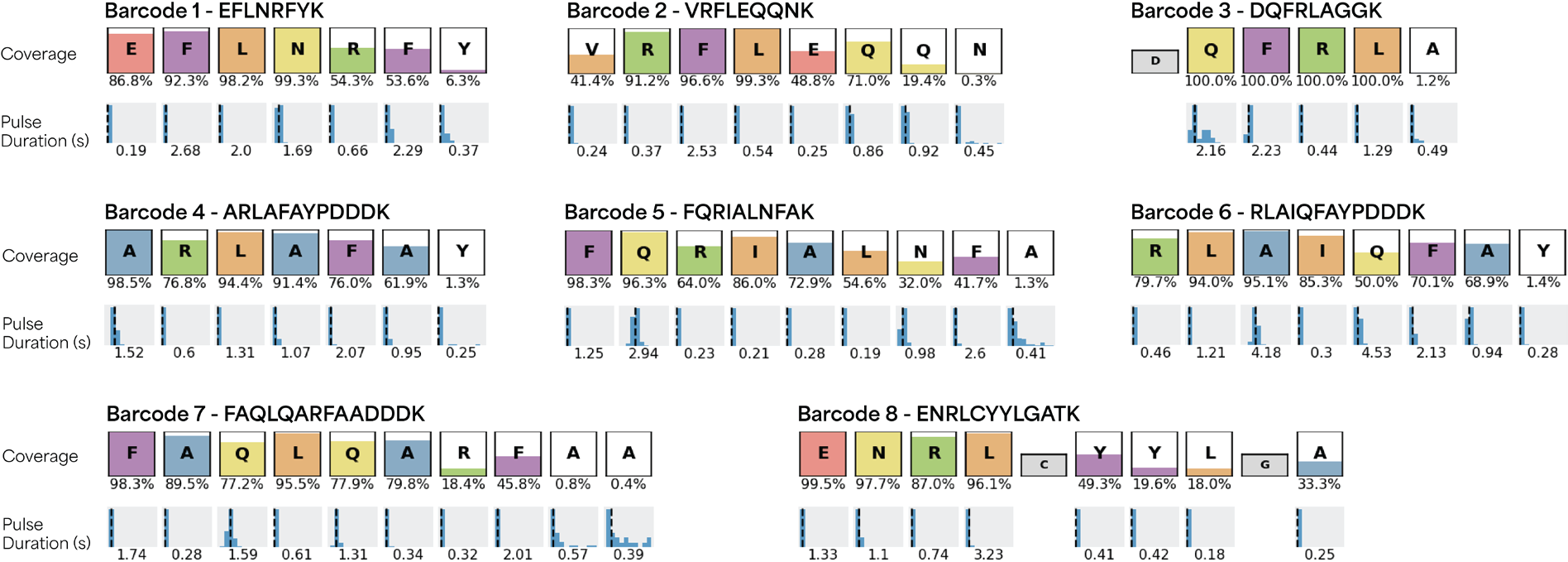
Figure 1. A set of peptide barcodes displaying unique patterns of RSs and on-chip pulse durations, enabling accurate identification with Platinum. The color fill in each box represents the total percent coverage of observed amino acids in each peptide sequence with the numerical value reported under each box. The mean pulse duration for each observed residue, measured in seconds, is given under each peptide sequence coverage box. Image Credit: Quantum-SI
The signals from each run were aligned to the entire set to measure the false discovery rate (FDR), which was defined as the fraction of off-target alignments for a particular peptide. The maximum FDR for all peptides was 0.2%, highlighting the strength of the NGPS in generating distinct RS patterns with kinetic properties and enabling the detection of each barcode with high confidence.
The Levenshtein distance (L) is a common measure of sequence similarity in bioinformatics defined as the minimum number of edit operations (insertions, deletions and substitutions) required to transform one sequence into another.8 The L was computed for every pair of sequences, and L ≥ 3 was observed for all pairs, with a mean L = 8.7 (Figure 2).
Combined with the distinct KSs and low FDRs observed in NGPS, this analysis implies that L ≥ 3 in all pairwise comparisons is an appropriate threshold to generate barcode sets with highly distinguishable sequences. The threshold was also utilized to evaluate how the sizes of PB sets could be increased.
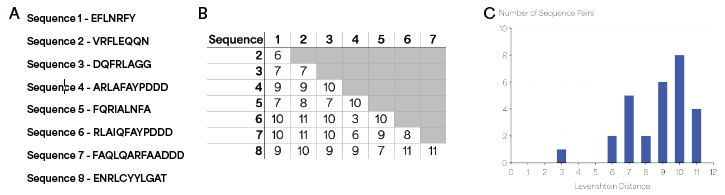
Figure 2. Levenshtein distance analysis of peptide sequences. (A) Sequences of peptides used in the Levenshtein distance analysis. (B) Levenshtein distance values for all pairs of sequences, showing L ≥ 3 for all pairs. (C) Histogram of orthogonal pairwise Levenshtein distances between all members of the barcode set. Image Credit: Quantum-SI
Many applications require the extension of a set to hundreds or thousands of sequences. To evaluate the scalability of PBs on Platinum, barcode sets were constructed in silico with L ≥ 3 for 6-amino-acid-character sets (L, F, R, N, A and E) which ranged in length from 3 to 8 residues. Repetitive sequences were restricted to ensure an amino acid could repeat only every five residues.
Barcode sets were created and ranged from 6-member sets for 3-residue barcodes to 1,600-member sets for 8-residue barcodes (Figure 3). Other design parameters, depending on the application, could be used to align with the desired throughput, accuracy and sensitivity.
These findings highlight the scalability of peptide design needed for applications requiring a large number of unique barcodes.
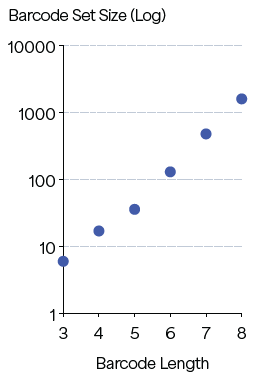
Figure 3. Predicted barcode set sizes at L ≥ 3 for barcode length of 3–8. Image Credit: Quantum-SI
Barcode fusing to functional elements, recombinant expression and mixture sequencing
To assess performance following translation, libraries were prepared from PBs expressed recombinantly in E. coli. The regular Quantum-Si library preparation workflow involves digesting samples with endoproteinase LysC, which produces peptides containing C-terminal lysine residues for conjugation to the K-linker.
Although this digestion process is necessary for proteomics workflows (where production of multiple peptides from the same protein is usually necessary for high-confidence target-identification) barcoding applications only require the recovery of a single barcoded peptide sequence for identification of the associated protein.
The digestion of samples containing multiple proteins or variants may lead to the production of an excess of non-barcoded peptides to barcoded ones. If both barcode and non-barcode peptides are then sequenced on Platinum, the detection sensitivity of the assay will be significantly reduced because the PBs will represent only a small portion of the available peptides for immobilization into the reaction chambers of the semiconductor chip.
An innovative solution to enrich and specifically conjugate the K-linker to the PB from a complex sample has been developed.
The novel approach uses a Sortase A enzyme-mediated transpeptidation reaction (Fig. 4A) which employs a two-step mechanism in which Sortase A first catalyzes the cleavage of a substrate (a short C-terminal LPETGG motif) between the threonine and glycine residues through a thioacyl intermediate. 9
The intermediate is then resolved via nucleophilic attack in trans from a glycine-containing substrate, which results in ligation via the formation of an amide bond. To incorporate this method into our process, we attached the LPETGG motif directly at the C-terminal of the peptide barcode sequence.
A Picolyl-Azide-Gly-Gly-Gly tripeptide was used to catalyze the required nucleophilic attack. This substrate also supplies the azide functional group for the subsequent click chemistry reaction to the K-linker.
The next task involves developing a method which specifically enriches and elutes the barcodes from a co-expressed protein of interest because digestion is no longer involved in the workflow. This was addressed by the incorporation of a SUMO tag at the N-terminus of the barcode sequence.
After the sample is enriched (by an affinity tag or a selection method) the tag enables the precise enzymatic cleavage of the barcode sequence from the protein coding sequence, using the SUMO protease ULP1, resulting in a free PB starting at the desired NAA of the peptide sequence.
In combination with the Sortase A transpeptidation approach, this method establishes an enzymatic preparation workflow which specifically elutes K-linker-conjugated PBs from enriched or selected samples, priming them for Platinum sequencing.
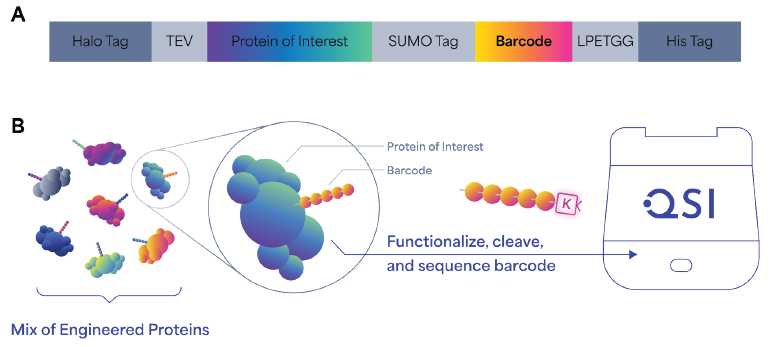
Figure 4. Design, generation, and sequencing workflow of barcoded protein libraries. (A) Schematic of barcode construct design, which includes a HaloTag for capturing on magnetic beads, TEV protease site to cleave the protein of interest with the barcode, a SUMO tag to specifically recover barcodes, a Sortase A recognition motif (LPETGG) for the enzymatic ligation of GGG-N3 peptide onto barcodes, and the His tag used for Ni-NTA/Talon purification of barcoded protein with all the tags. (B) Schematic representation of the generation of barcoded protein libraries. Barcoded protein libraries are expressed in vitro or in vivo and subject to screening or selection. Barcodes are then cleaved and conjugated to K-linker before sequencing on Platinum. Image Credit: Quantum-SI
The fully enzymatic library preparation workflow was tested on barcodes recombinantly expressed in E. coli, with the aim to establish that these barcodes can be identified with a minimal FDR in a controlled mixture of PBs with varying ratios. An 8-PB mixture library, in which each barcode was added to the mixture at the indicated amounts shown in Table 1, was prepared and sequenced.
As Figure 5 shows, each of the barcodes was identified in the mixture using NGPS, and the number of alignments deceased with relative abundance. Out of eight peptides, seven yielded FDR values of < 10 %. Of these peptides, six yielded FDR values of < 1 %, and one ~5 %, demonstrating the capability of PBs to endure recombinant expression, purification and sequencing using Platinum.
Table 1. Relative abundance of barcodes. Source: Quantum-SI
| |
Relative Abundance |
| Barcode 6 |
1x |
| Barcode 1 |
0.67x |
| Barcode 7 |
0.67x |
| Barcode 2 |
0.67x |
| Barcode 5 |
0.5x |
| Barcode 4 |
0.5x |
| Barcode 3 |
0.5x |
| Barcode 8 |
0.1x |
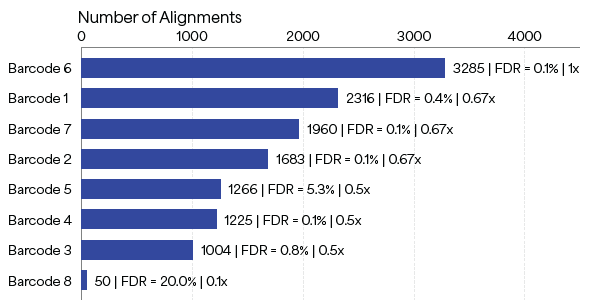
Figure 5. Sequencing results on Platinum of an 8-peptide barcode mixture library. Each peptide barcode was added to the mixture at controlled relative amounts. The metrics at the end of the bars represent Number of Alignments | FDR | Relative Abundance. For example, Barcode 1 was added to the mixture at a relative abundance of 0.67x, yielding 2,316 alignments with an FDR of 0.4%. Image Credit: Quantum-SI
Single-molecule peptide sequencing for nanobody enrichment screening
Nanobodies (single-domain camelid antibodies, which have gained eminence in various applications, such as targeted drug therapy) were used to demonstrate the application of PBs in a differential enrichment assay.
Nanobodies can be chosen from large synthetic libraries using high-throughput approaches, such as phage or ribosome display, but such methods are limited because of the constraint to maintain phenotype and genotype linkage. Once this requirement is removed, barcodes offer an efficient solution for screening variants.
Here, a model library with two nanobodies was used: an anti-MBP nanobody (targeting maltose binding protein or MBP) and an anti-GFP nanobody (targeting green fluorescent protein, GFP).6,7 At the C-terminus, the nanobodies were fused with the MBP PB and GFP PB, respectively (Figure 6).
The nanobodies were prepared for sequencing based on the targeted sequencing approach described in the previous section. The barcoded nanobody library was added to a solution with magnetic beads coated with GFP throughout the affinity selection process.
Successively, the PBs were proteolytically eluted from the nanobodies bound to the beads. The post-selection library was then sequenced using Platinum alongside a pre-selection control library. The relative number of each barcode alignment pre- and post-selection were compared.
As seen in the graph in Figure 6, the proportion of GFP to MBP PB alignments was approximately 3.18:1 in the pre-selection control library, increasing to 1079:1 in the post-selection library, suggesting enrichment of approximately 300-fold of the anti-GFP nanobody. This indicates the ability of the method to measure the differential enrichment of nanobody clones in a library through direct single-molecule sequencing of protein-associated barcodes.
The results also reveal the advantage of targeted sequencing in specific applications. Shotgun sequencing of these two nanobodies would have produced 11 individual digested peptides: 9 from the two nanobodies plus the 2 barcodes.
Conversely, the targeted sequencing approach (which increases barcode sequencing capacity by approximately 5-fold) was applied to two small (15 kDa each) proteins; this effect would be even more pronounced in applications targeting larger proteins of interest, in which shotgun sequencing would produce considerably more peptides.
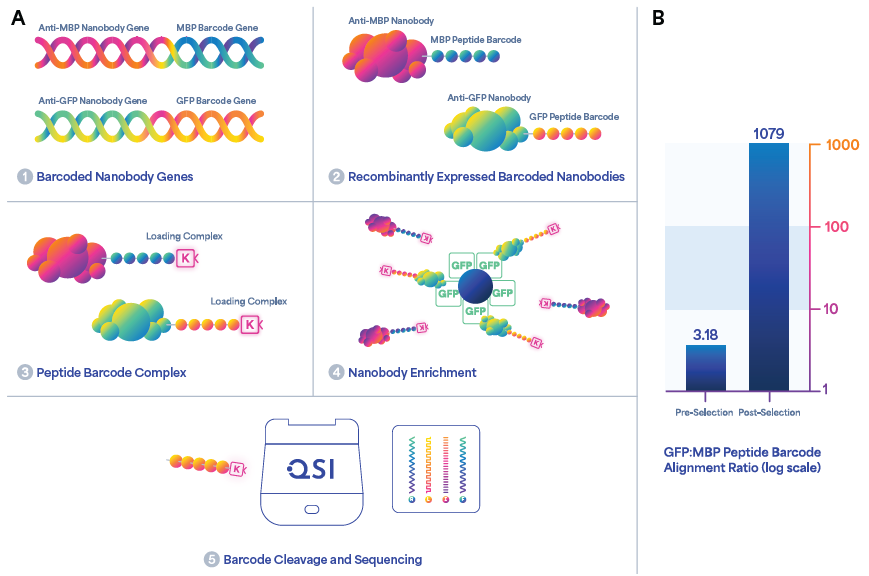
Figure 6. Workflow for enrichment and sequencing of barcoded nanobodies. (A) Schematic representation of the differential enrichment of an anti-GFP nanobody by peptide barcoding. Model anti-GFP and anti-MBP nanobody genes were encoded with unique peptide barcodes and expressed recombinantly. The proteins were purified, labeled with an azide tag, and conjugated to macromolecular linkers. The nanobodies were enriched using GFP immobilized on magnetic beads. After selection, the barcodes were eluted by proteolysis and sequenced on Platinum. (B) Bar graph quantification of pre- and post-selection barcoded nanobody libraries indicating a > 300-fold enrichment for the anti-GFP nanobody post-selection based on the ratio of GFP to MBP peptide barcode alignments. Image Credit: Quantum-SI
Conclusion
The utilization of PBs in fields, including synthetic biology, proteomics, drug discovery and protein engineering, is promising. Research suggests that short peptide sequences yield rich information and distinctive KSs which can be recognized accurately.
The optimized barcode designs also guarantee reliable relative quantification. Furthermore, introducing an enzymatic approach to targeted sequencing allows the easy and precise retrieval of barcodes from complex matrices, such as E. coli lysate, which will be crucial in future work.
References and further reading
- Wang, Y. and Navin, N. E. (2015) Advances and Applications of Single-Cell Sequencing Technologies. Mol. Cell 58, pp.598–609
- Whitmann, B. J., Johnston, K. E., Almhjell, P. J. and Arnold, F. H. (2022) evSeq: Cost-Effective Amplicon Sequencing of Every Variant in a Protein Library. ACS Synth Biol 11, pp.1313–1324.
- Baysoy, A., Bai, Z., Satija, R. and Fan, R. (2023) The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell Biol. 24, pp.695–713.
- Egloff, P. et al. (2019) Engineered PBs for In-Depth Analyses of Binding Protein Ensembles. Nat. Methods 16, pp.421–428.
- Reed, B. D. et al. (2022) Real-time dynamic single-molecule protein sequencing on an integrated semiconductor device. Science 378, pp.186–192.
- Fridy, P. C. et al. (2014) A robust pipeline for rapid production of versatile nanobody repertoires. Nat. Methods 11, pp.1253–1260.
- Zimmermann, I. et al. (2018) Synthetic single domain antibodies for the conformational trapping of membrane proteins. eLife 7, e34317.
- Levenshtein, V. I. (1965) Binary codes capable of correcting deletions, insertions, and reversals. Dokl. Phys. 163, pp.845–848.
- Mazmanian, S. K., Ton-That, H. and Schneewind, O. (2001) Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 40, pp.1049–1057.
- Jovčevska, I. and Muyldermans, S. (2020) The Therapeutic Potential of Nanobodies. BioDrugs 34, pp.11–26.
About Quantum-SI
Inspired by Ion Torrent’s success at shrinking next-generation sequencing technology into a benchtop instrument, Jonathan Rothberg founded Quantum-Si™ to bring the same semiconductor technology to protein sequencing with the launch of the Platinum® Next-Generation Protein Sequencer™.
That was in Guilford, CT, back in 2013. Fast forward to today and we now have over 1,000 patents issued and applications pending, plus a groundbreaking single-molecule protein sequencing technology platform, the Platinum.
Along the way, we solved critical challenges around sensitive and unambiguous amino acid detection, blending biology, chemistry, and semiconductor technology to help biologists see what other approaches cannot deliver. We also set the stage for a revolution in how scientists understand biology and build new treatments for disease by making single molecule protein sequencing accessible to every lab everywhere.
We are now entering a new phase of our development as a company. Starting with an initial public offering in June 2021 (QSI on the NASDAQ) and continuing with a new product development and operations facility in San Diego, CA, in 2022, we have entered a period of rapid growth. Through this expansion, we will be able to fuel a new era of biology, the post-genomic era, where biologists accelerate basic scientific insight and biomedical advances through the power of next-generation protein sequencing.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.