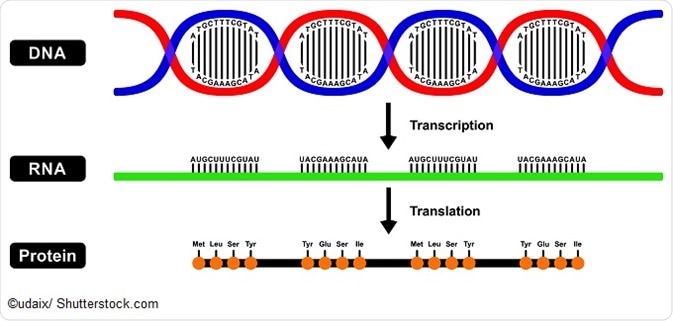
Each protein or peptide consists of a linear sequence of amino acids. The protein primary structure conventionally begins at the amino-terminal (N) end and continues until the carboxyl-terminal (C) end. The structure of a protein may be directly sequenced or inferred from the sequence of DNA.
The amino acid sequence of a protein or peptide is useful information to understand the protein or peptide, identify it in a sample and categorize its post-translational modifications. The process of determining the amino acid sequence is known as protein sequencing.
Notation
The sequence of a protein is usually notated as a string of letters, according to the order of the amino acids from the amino-terminal to the carboxyl-terminal of the protein. Either a single or three-letter code may be used to represent each amino acid in the sequence.
There are 20 amino acids that occur naturally in nature, which can be represented by a three or single letter code as follows:
- Alanine (Ala, A)
- Arginine (Arg, R)
- Asparagine (Asn, N)
- Aspartic acid (Asp, D)
- Cysteine (Cys, C)
- Glutamic acid (Glu, E)
- Glutamine (Gln, Q)
- Glycine (Gly, G)
- Histidine (His, H)
- Isoleucine (Ile, I)
- Leucine (Leu, L)
- Lysine (Lys, K)
- Methionine (Met, M)
- Phenylalanine (Phe, F)
- Proline (Pro, P)
- Serine (Ser, S)
- Threonine (Thr, T)
- Tryptophan (Trp, W)
- Tyrosine (Tyr, Y)
- Valine (Val, V)
Methods of Protein Sequencing
There are two main methods used to find the amino acid sequences of proteins. Mass spectrometry is the most common method in use today because of its ease of use. Edman degradation using a protein sequenator is the second method, which is most useful if the N-terminus of a protein needs to be characterized.
It is helpful to know which amino acid is at the N-terminus of the protein both for ordering of the peptide fragments into the whole chain and to reduce the impact of impurities that commonly occur in the first round of Edman degradation. The N-terminus can be identified by:
- Using a reagent to label the amino acid at the end of the protein.
- Hydrolyzing the protein
- Using chromatography and other methods of comparison to identify the marked protein.
There are fewer methods that can practically be used to identify the C-terminus of the protein. However, one method that may be used involves adding carboxypeptidases to a solution of the protein and taking regular samples. Plotting the concentration of amino acids against time can help to identify the amino acid at the C-terminus.
Edman degradation allows the sequence of amino acids in the protein to be discovered with Edman sequencers, which are currently able to sequence peptides up to about 50 amino acids in length. This involves several steps to:
- Use a reducing agent to break any disulfide bridges in the protein.
- Separate the chain(s) of the protein complex and purify them.
- Determine the composition and terminal amino acids of each chain.
- Break each chain into small fragments (less than 50 amino acids in each)
- Separate the fragments and purify them.
- Use the fragments to determine amino acid sequence.
- The preceding steps should be repeated with a different fragment pattern so that the overall protein sequence can be reconstructed with minimal errors.
Amino Acid Composition and Analysis
The unordered composition of an amino acid is often useful information when attempting to determine the ordered sequence of the protein. This is because it can help identify errors and interpret ambiguous results. Additionally, the frequency of amino acids can also help to decide upon the protease that is more appropriate for the protein digestion.
There are two main steps to determine the frequency of amino acids in a process known as amino acid analysis. Firstly, hydrolysis of a known quantity of the protein should break it up into the amino acid monomers. These can then be separated and quantified using various methods.
The hydrolysis is typically carried out by heating a sample of the protein to over 100°C in hydrochloric acid for an extended period of time (at least 24 hours), allowing more time for proteins with bulky hydrophobic groups. As there is a risk of protein degradation in these conditions, particularly for cysteine, glutamine, serine, threonine, tryptophan, and tyrosine, it is recommended to use several samples and to heat them for different times. Once hydrolyzed, the amino acids can be separated and identified with techniques such as ion-exchange chromatography or reverse phase HPLC.
References
- https://www.ncbi.nlm.nih.gov/
- https://www.youtube.com/watch?v=iACY379o1X4
Further Reading
Last Updated: Jul 19, 2023
 Exploring Targeted Protein Degradation as a New Strategy to Tackle Antimicrobial Resistance
Exploring Targeted Protein Degradation as a New Strategy to Tackle Antimicrobial Resistance