There has been much controversy over the contribution of various host gene variants to the severity of the coronavirus disease 2019 (COVID-19) in different individuals. A new preprint on the preprint server medRxiv* discusses the importance of the common, rare, and intermediate variants of several host genes to the clinical severity of disease following infection with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
The researchers of the current study aimed to develop a model to predict the severity of COVID-19 using the host genes, defining severe disease when patients were hospitalized or required respiratory support in any form. The model was designed to be both reliable and easily interpreted by clinical staff, helping in the rapid triage of patients who might require oxygen support early to forestall the development of further complications.
The challenge was to make sense of the vast number and diversity of host genes to prove a direct link between a gene variant and disease severity. To do this, a large number of variants must be compared against individual patients who are far fewer in number. This process is often considered to be too complicated for the findings to be reliable.
The current study, therefore, used gene-level representation with a machine-learning algorithm. The algorithm is based on the premise that while both common and rare variants contribute to disease severity, they do so to varying extents.
That is, one rare variant that leads to aberrant protein function may be adequate for severe disease, whereas commonly occurring variants are less unlikely to affect protein function. The researchers, therefore, developed a score known as the Integrated PolyGenic Score (IPGS), which incorporates data on the variants at different frequencies.
Training the algorithm
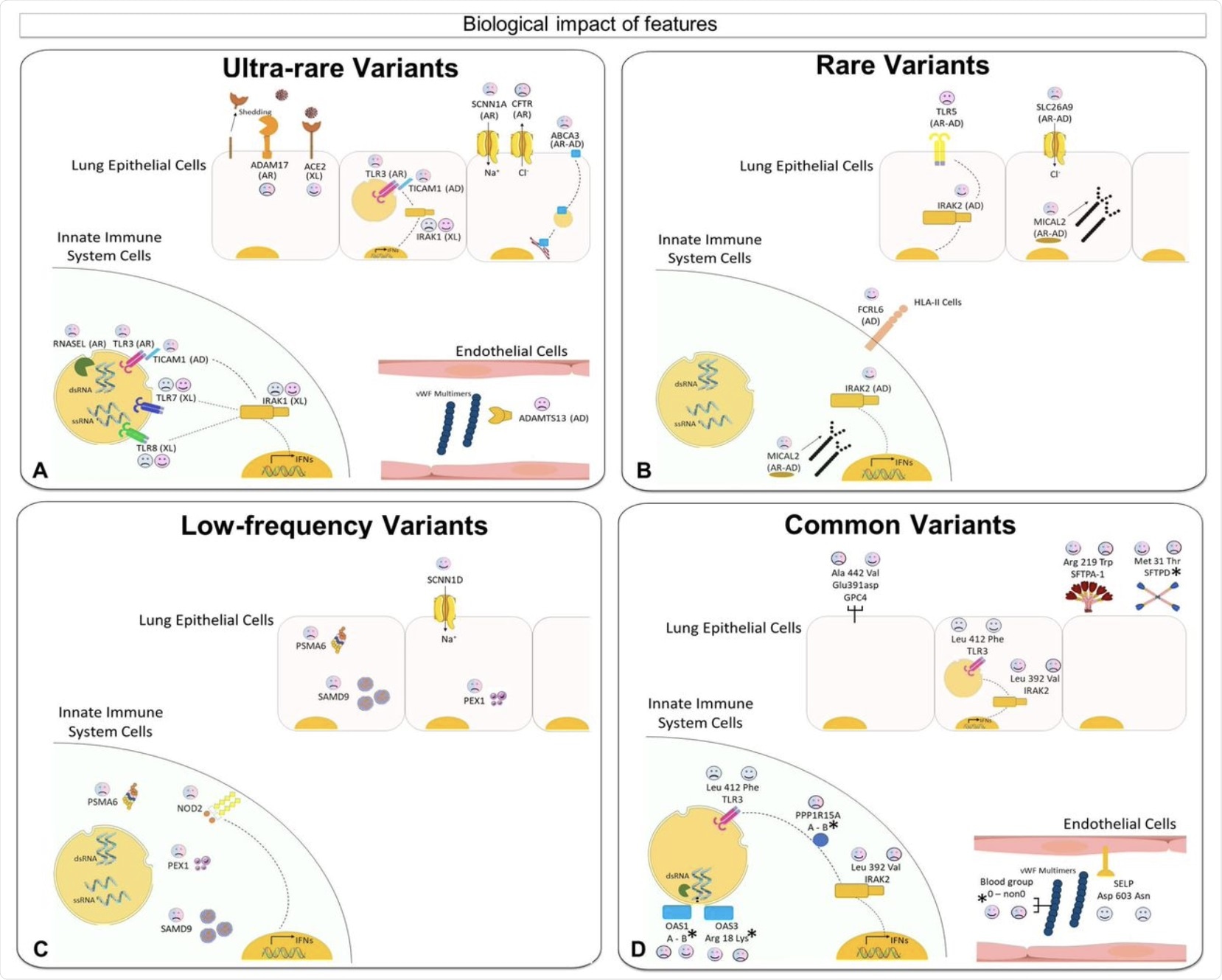
The data was taken from a dataset called GEN-COVID, which included approximately 1,800 patients. Phenotypic features were processed into 12 sets, such as “ultra-rare_autosomal dominant” (UR_AD), “ultra-rare_autosomal recessive” (UR_AR), as well as “ultra-rare_X-linked” (UR_X). Each of these features was designed to represent various autosomal dominant and recessive models of inheritance.
The researchers of the current study also reduced the number of input features using a feature selection strategy with Least Absolute Shrinkage and Selection Operator (LASSO) regularization.
Over half of the genes were found to be contributed by a single variant, whereas approximately 30% contributed by two, 11% by three, and 6% by four. About 25% of the genes contributed in a sex-specific manner. These were either on the X chromosome or were regulated in contrasting directions by androgens and estrogens.
Some genes have been previously found to be implicated in COVID-19 in a Mendelian-like fashion. Others offer a high chance of being direct contributors because of their functional involvement in the disease, such as those involved in innate immunity, coagulation, or ADAM17, which sheds the angiotensin-converting enzyme 2 (ACE2), which is the receptor used by SARS-CoV-2 to gain entry. Into the host
Some rare variants included inflammatory genes like TLR5 and SLC26A9.
What did the model show?
The model performed better when IPGS was included compared to age and sex alone, showing a clear difference between severe cases and others for both the male and female cohorts.
Using several testing groups, the accuracy and precision of the prediction improved by 1.3% and 1%, respectively, with a 1.3% improvement in sensitivity and 1.67% enhancement of specificity. When adjusted for the presence of other illnesses, the odds for accurate prediction were 2.5 times higher, thus confirming that this score is reliable in providing a prognosis of severe disease.
Compared with other sex/age models, or a combined model, the IPGS predicted severity 2.3 times better. IPGS was also found to offers biological plausibility and the potential for a better understanding of how the disease develops, as well as how it can be treated.
In three male patients who had a severe outcome, the IPGS model indicated a probability of severity of 0.91 to 0.95, while the age/sex model only showed 0.52 to 0.66. The gene variants that may be of therapeutic importance include the TLR7 ultra-rare mutation, which may respond to gamma-interferon; homozygous 603Asn variant in the SELP gene, which may indicate adjuvant treatment with anti-selectin P autoantibodies; and polyQ chain of more than 23 Q residues in the AR gene, which may respond to testosterone.
A female patient was also correctly predicted to be at risk of severe disease despite being in her early thirties, as a result of the presence of the ADAMTS13 ultra-rare mutation. The presence of this mutation increases the risk of thrombosis as a result of reduced von Willebrand factor cleavage.
Two other elderly male patients with mild outcomes had IPGS scores of only 0.23 and 0.41. These individuals exhibited ultra-rare ACE2 mutations that may have contributed to the reduced viral load, as well as AGTR2 mutations, which are linked to reductions in the lung manifestations of cystic fibrosis.
Implications
As in other disorders with a complex etiology, both rare and less common genetic variants contribute to the susceptibility as well as the severity of COVID-19. The latter is demonstrated in the current study.
The researchers also showed that while one ultra-rare mutation could cause severe COVID-19 by itself, this is a less frequent outcome with a common variant. They also discovered that ultra-rare, rare, low-frequency and common variants should be considered separately in order to extract all types of genes effectively.
The IPGS model may thus help to provide a reliable prediction of the chances of severe disease after SARS-CoV-2 infection. This model has certain advantages over the older Polygenic Risk Score (PRS), such as including only coding variants and giving differential weights to each feature relative to the frequency, which is likely related to the effect on the protein.
Each patient receives a list of the common and less frequent polymorphisms that may impact COVID-19 outcomes. This could help select personalized adjuvant therapy.
“At the time of writing, a platform trial based on genetic markers is being discussed with the Italian Medicines Agency.”
The identification of such risk polymorphisms could help identify the linked gene variants that are associated with greater susceptibility and/or severity. An interesting example is the TLR7/8 gene on the X chromosome.
Females have twice as much of this protein as males because of their two X chromosomes. Comparatively, males with an ultra-rare TLR7/8 mutation develop severe disease, whereas females fare better than those without the mutation.
“The activation of TLR7/8 induces the production of type 1 and type 2 IFN as well as pro-inflammatory cytokines, where the production defect in hemizygous males leads to severe COVID-19. However, an excess of the sensor can also lead to damage from hyperinflammation. Therefore, the condition of carrier females is the more favorable state and has in fact been associated with mild COVID-19.”
The results also point to relevant pathways in COVID-19 pathogenesis, including ciliary motility, and clathrin-mediated endocytosis. Thus, IPGS is a novel prognostic score that should be exploited when treating COVID-19 patients.
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Fallerini, C., Picchiotti, N., Baldassarri, M., et al. (2021). Common, Low-Frequency, Rare, And Ultra-Rare Coding Variants Contribute To COVID-19 Severity. medRxiv. doi:10.1101/2021.09.03.21262611. https://www.medrxiv.org/content/10.1101/2021.09.03.21262611v1
- Peer reviewed and published scientific report.
Fallerini, Chiara, Nicola Picchiotti, Margherita Baldassarri, Kristina Zguro, Sergio Daga, Francesca Fava, Elisa Benetti, et al. 2021. “Common, Low-Frequency, Rare, and Ultra-Rare Coding Variants Contribute to COVID-19 Severity.” Human Genetics, December. https://doi.org/10.1007/s00439-021-02397-7. https://link.springer.com/article/10.1007/s00439-021-02397-7.
 Gene therapy targeting connexin 43 shows promise across inherited heart diseases
Gene therapy targeting connexin 43 shows promise across inherited heart diseases