Since the onset of the COVID-19 pandemic, several severe acute respiratory syndrome coronavirus 2 (SARS‑CoV‑2) variants of concern (VOC) have emerged, leading to repeated surges in cases, deaths, and hospitalizations throughout the world. Classification of these variants by the Phylogenetic Assignment of Named Global Outbreak Lineages (PANGO) nomenclature shows that although they have descended from a common ancestor, they are not direct descendants of one another.
The PANGO lineages that have been corresponded to the VOCs include Alpha variant (B.1.1.7 and Q lineages), Beta variant (B.1.351 and descendant lineages), Gamma variant (P.1, which is a descendant of B.1.1.28, and descendant lineages), Delta variant (B.1.617.2 and AY lineages), and Omicron variant (B.1.1.529 and BA lineages).
All the variants were reported to have evolved from the B.1 lineage, while Alpha, Gamma, and Omicron also have B.1.1 as an additional parent lineage. However, these classifications do not describe the degree of distinctiveness between the variants or provide insights into the genetic properties of the variants.
The evolution of SARS-CoV-2, like all other viruses, occurs via the mutation of its genome; these mutations alter the amino acid sequences of the viral proteins. The mutations can be either positively or negatively selected based on their impact on viral fitness. Mutations in several regions, such as the N-terminal domain (NTD) of the Spike glycoprotein and receptor-binding domain (RBD), improved viral fitness. Although much attention has been given to individual mutations at the amino acid level, limited attention has been given to the nucleotide sequence level.
A new study published in the pre-print server medRxiv* hypothesized that the emergence of more immune invasive or transmissible variants of SARS-CoV-2 was associated with increased genetic distinctiveness from the original or previous strains.
 Study: Genomic diversification of long polynucleotide fragments is a signature of emerging SARS-CoV-2 variants of concern. Image Credit: NIAID
Study: Genomic diversification of long polynucleotide fragments is a signature of emerging SARS-CoV-2 variants of concern. Image Credit: NIAID
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
To test the hypothesis, the study introduced a new methodology that quantifies the number of distinct nucleotide n-mers (of various sizes) in VOCs to estimate the degree of viral evolution.
About the study
The study involved calculating and quantifying the number of distinctive n-mers for SARS-CoV-2 sequences from the original reference strain (PANGO lineage A) and five VOCs, Alpha, Beta, Gamma, Delta, and Omicron, that were obtained from the GISAID database. In addition, the number of amino acid mutations for the sequences obtained from GISAID were determined and compared to the original Wuhan-Hu-1 strain of SARS-CoV-2.
Multiple sequence alignment (MSA) was carried out for the sub-sampled SARS-CoV-2 genomes to calculate the phylogenetic distance. Finally, the distinctiveness of n-mers for a specific SARSCoV-2 lineage was calculated using an alternative metric, A*(1-B).
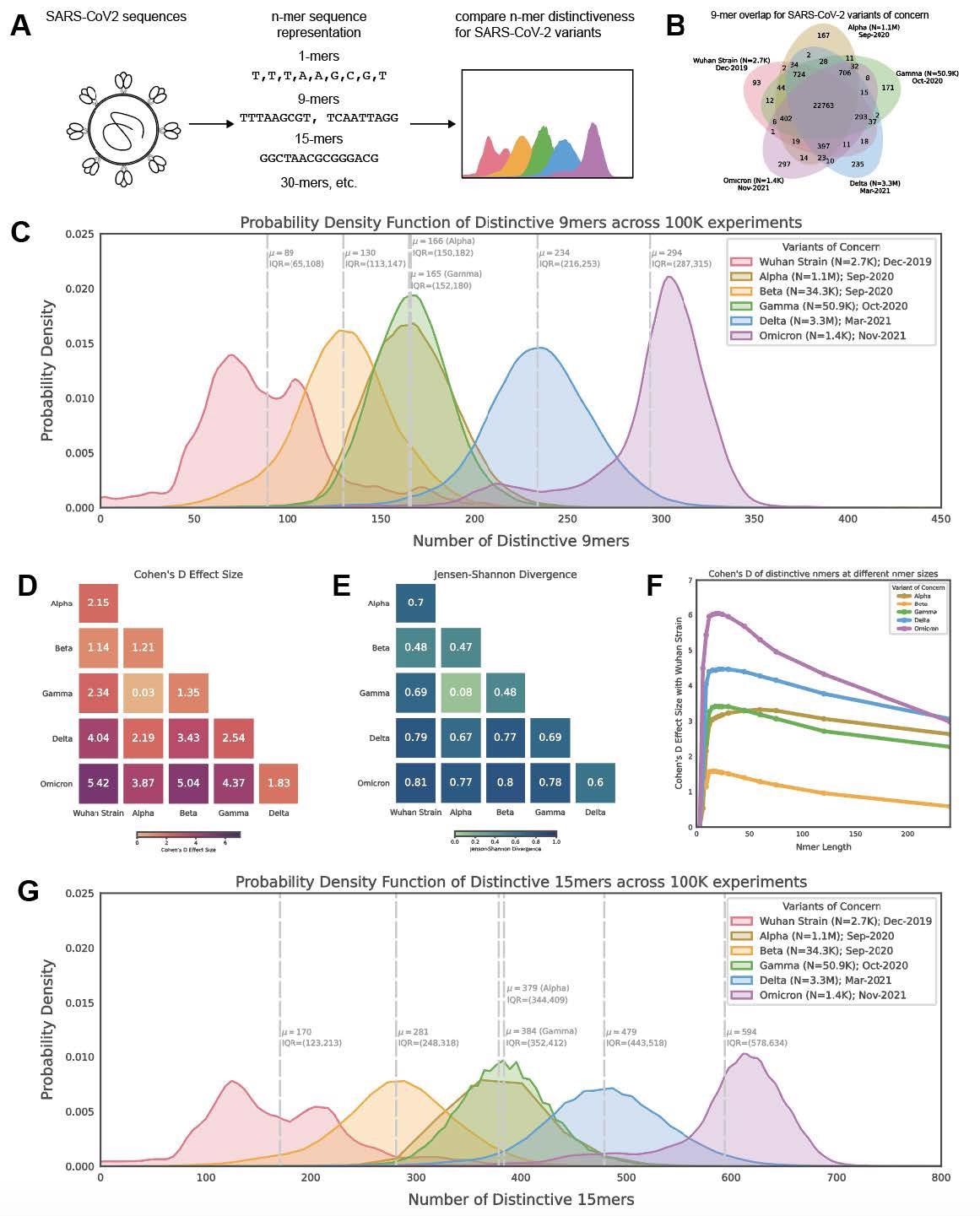
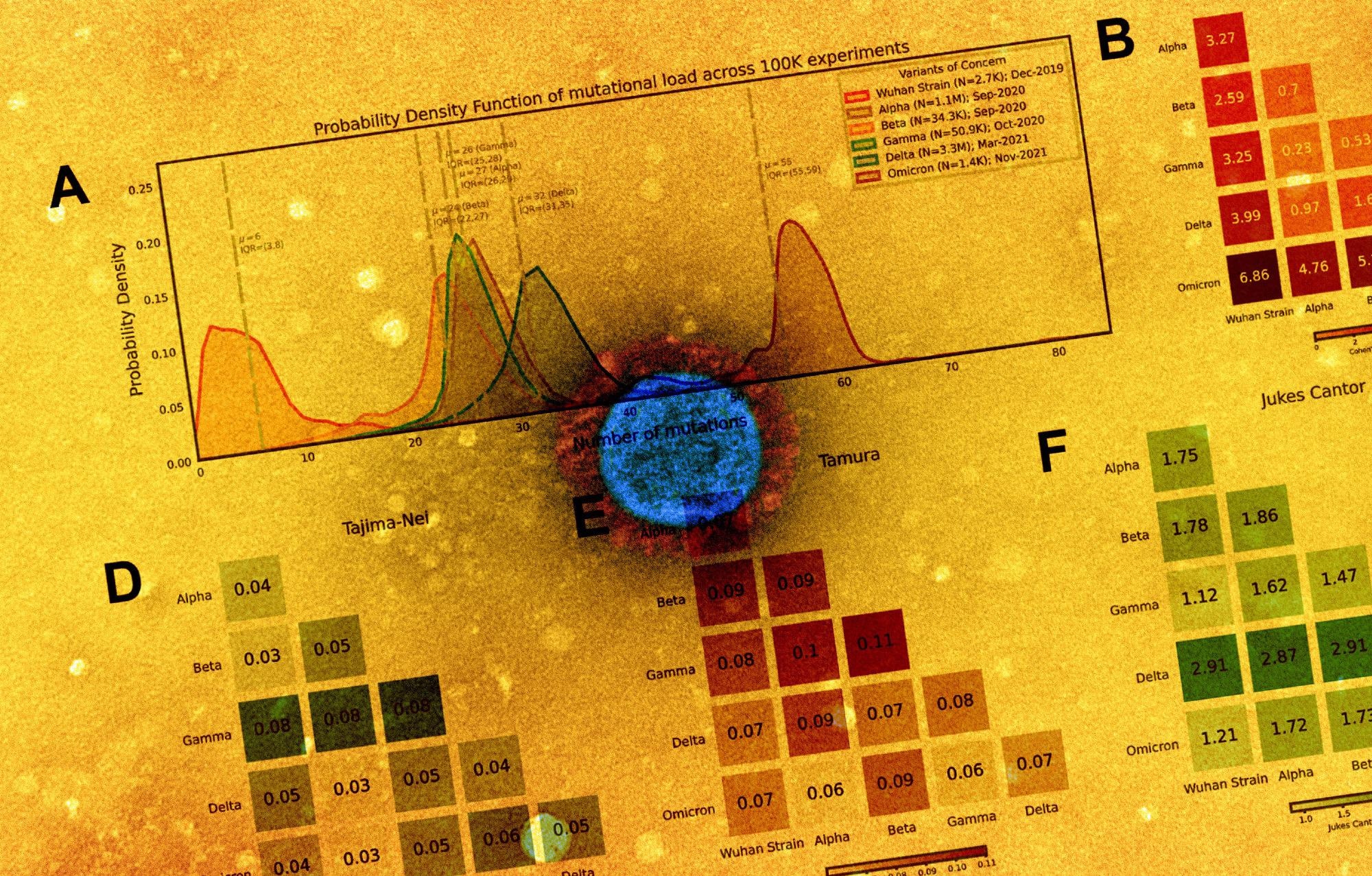
Distribution of polynucleotide distinctiveness for SARS-CoV-2 variants of concern (VOCs). (A) Schematic illustration of polynucleotide sequence analysis. SARS-CoV-2 sequences are analyzed to generate a set of distinct n-mer polynucleotide sequences (max n-mer size = 240). (B) Venn Diagram showing the mean of the distributions for shared and unique nucleotide 9-mers between all combinations of variants across 100,000 replicate comparisons. The Beta variant was excluded from this visualization to reduce clutter. (C) Density plots showing 9-mer sequence distinctiveness for VOCs, as measured by the number of distinct 9-mer polynucleotide sequences. (D-E) Heatmaps showing Cohen’s D and Jensen-Shannon divergence values from pairwise comparisons of the distributions shown in (C). (F) Cohen’s D of the distinctive n-mer distributions of Alpha, Beta, Gamma, Delta, and Omicron variants against the original strain for various n-mer lengths (n = 3, 6, 9, 12, 15, 18, 21, 24, 30, 45, 60, 75, 120, and 240). (G) Density plots showing an additional example for genomic distinctiveness of VOCs, as measured by the number of distinct 15-mer polynucleotide sequences. Data shown in panels B-G were generated using 287,739 unique SARS-CoV-2 sequences in total, split across the variants as shown in the legend of C. Abbreviations: μ - mean; IQR - interquartile range; VOC - variant of concern.
Study findings
The results reported that from each genome, a distinctive nucleotide 9-mers (DN9s) were derived that was present in a given lineage but absent from all others. The number of DN9s corresponded to the time of emergence and was found to be highest for Omicron, followed by Delta, Alpha, Gamma, and finally Beta variant. The Omicron sequence was also found to have more DN9s than all other VOCs.
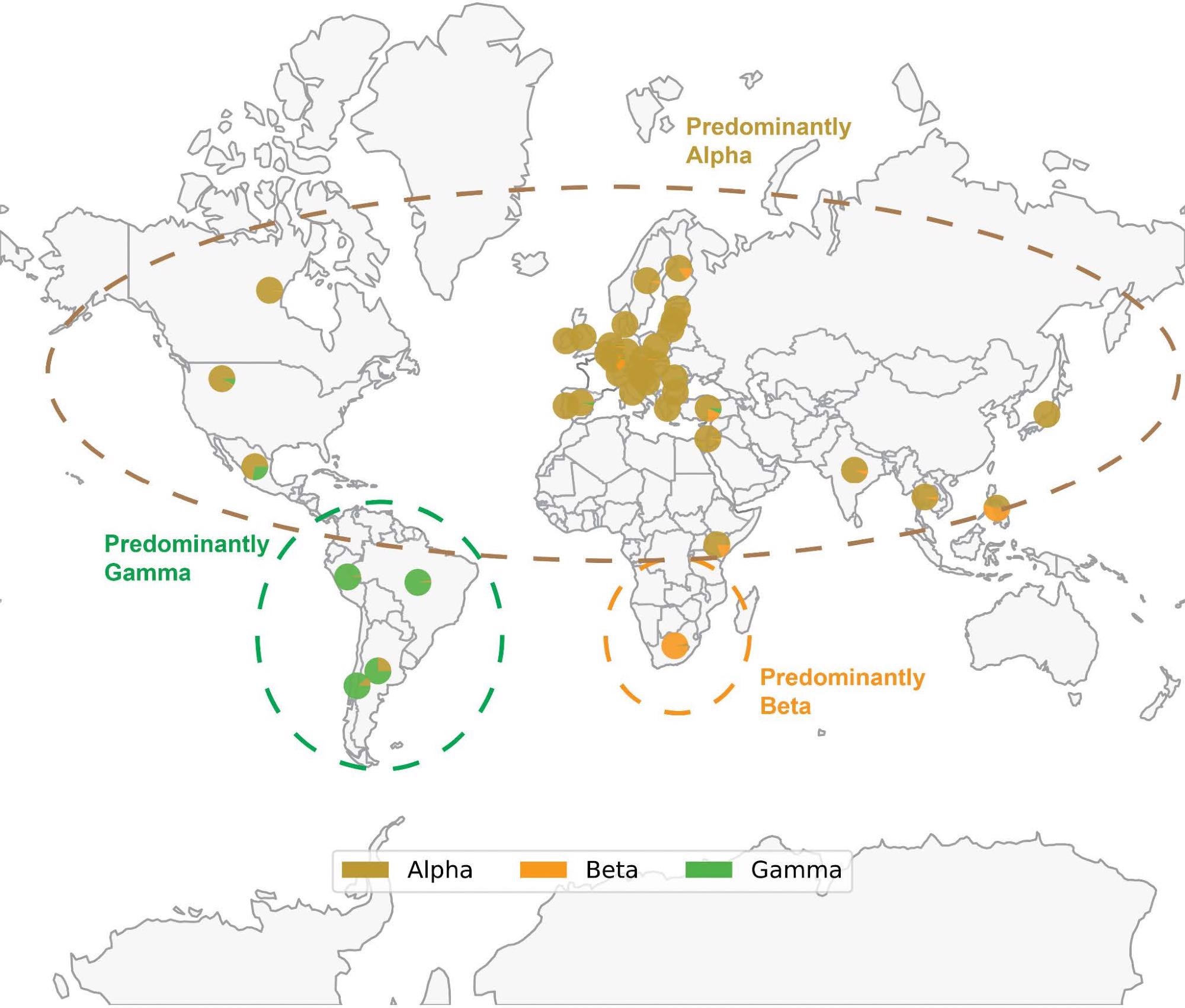 Map of SARS-CoV-2 VOC prevalence by geographic region. Geographical distribution of Alpha (B.1.1.7), Beta (B.1.351) and Gamma (P.1) variants based on sequences deposited in GISAID through December 14, 2021. Each pie chart shows the proportion of Alpha, Beta or Gamma sequences deposited in the country. Note that the denominator is the number of sequences labeled as any of these three variants, rather than the total number of sequences deposited in that country. Thus, each pie chart answers the following question: “Of all genomes deposited in a given country which were assigned as Alpha, Beta, or Gamma, what proportion of genomes was assigned to each of these three lineages?” The prevalence of Delta and Omicron are not shown to better highlight the geographical distribution of Alpha, Beta, and Gamma; however, Delta and Omicron are currently or have previously been highly prevalent in the regions shown. Only countries where at least 1000 sequences are deposited are shown. The variants depicted, which circulated at approximately the same time, generally became prominent in geographically distinct regions.
Map of SARS-CoV-2 VOC prevalence by geographic region. Geographical distribution of Alpha (B.1.1.7), Beta (B.1.351) and Gamma (P.1) variants based on sequences deposited in GISAID through December 14, 2021. Each pie chart shows the proportion of Alpha, Beta or Gamma sequences deposited in the country. Note that the denominator is the number of sequences labeled as any of these three variants, rather than the total number of sequences deposited in that country. Thus, each pie chart answers the following question: “Of all genomes deposited in a given country which were assigned as Alpha, Beta, or Gamma, what proportion of genomes was assigned to each of these three lineages?” The prevalence of Delta and Omicron are not shown to better highlight the geographical distribution of Alpha, Beta, and Gamma; however, Delta and Omicron are currently or have previously been highly prevalent in the regions shown. Only countries where at least 1000 sequences are deposited are shown. The variants depicted, which circulated at approximately the same time, generally became prominent in geographically distinct regions.
Omicron was indicated to be the most highly mutated VOC, while the phylogenetic distance between Gamma from Alpha and Beta was the most notable. The results also suggest that the newly emerging SARS-CoV-2 variants were genetically distinct from the original strain and that they comprised unique nucleotide sequences that resulted in the distinctiveness. The distinctiveness was also found to increase within a lineage with evolutionary time.
The current study thus provides a new methodology that will help the researchers identify and assess the distinctiveness of any new SARS-CoV-2 variants compared to the previous ones. However, further research is required to determine whether this method will be able to classify lineages as VOCs earlier than the time taken currently, how vaccination would impact the SARS-CoV-2 genomic diversity, and also determine whether SAR-CoV-2 infection would progress towards seasonality or endemicity.
Limitations
The study had certain limitations. First, since the number of Omicron sequences available in the GISAID database is currently low, it can lead to oversampling. Second, apart from nucleotide 9-mers, protein-coding nucleotide n-mers or amino acid n-mers should also be considered in the determination of genomic diversity. Third, the study can be sensitive to the lineage composition in the complement group. Finally, further research is required regarding the relationship between genomic distinctiveness metrics with phylogenetic depth and evolutionary time.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
 Poor lung gas exchange may help explain lingering brain symptoms in Long COVID
Poor lung gas exchange may help explain lingering brain symptoms in Long COVID