The risk and clinical outcomes of SARS-CoV-2 infection vary widely among individuals. However, the mechanisms underlying these differences in coronavirus disease 2019 (COVID-19) progression and pathogenesis remain unclear. Studies have shown that the mucosal microbiome influences host toll-like receptor (TLR) expression, which is involved in virus detection. Studies have also shown the prevalence of bacterial co-infections in COVID-19 patients at 6.9%.
Likewise, elevated nasal cytokines have been implicated in adverse clinical outcomes in influenza patients. Therefore, exploring upper respiratory tract microbiota compositional changes following SARS-CoV-2 infection could help evaluate its impact on infection severity.
About the study
In the present study, researchers performed Nanopore full-length 16S ribosomal ribonucleic acid (rRNA) sequencing on 194 nasopharyngeal swab specimens collected between March 2020 and January 2022 from the general population of British Columbia, Canada. They constituted four study groups comprised of hospitalized (HOSPOS) COVID-19 patients, community-dwelling (COMPOS) COVID-19 patients, hospitalized SARS-CoV-2-uninfected patients (HOSNEG), and uninfected community dwellers (COMNEG).
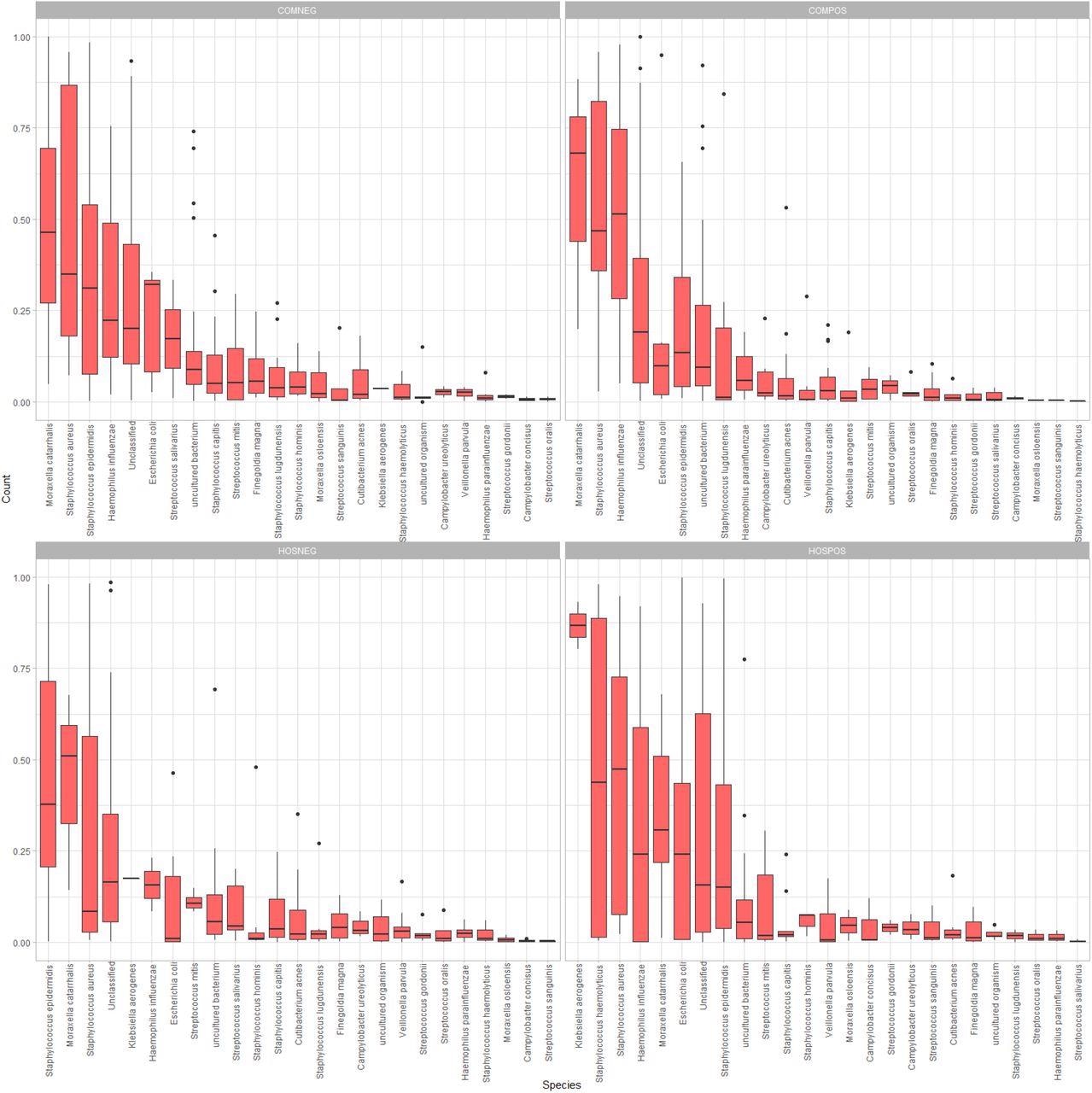
Side-by-side boxplots of relative abundance at the species-level among our four study groups.
Further, the team performed sequence data analysis using the BugSeq 16S analysis pipeline. They performed routine diagnostic testing and a lab-developed (LDT) real-time reverse transcriptase-polymerase chain reaction (RT-PCR) assay on the study samples to detect SARS-CoV-2 envelope (E) and RNA-dependent RNA polymerase (RdRp) gene targets. They also used an in-house developed PCR-based assay to screen the SARS-CoV-2 variants of concern (VOCs), Alpha, Beta, Gamma, Delta, and Omicron in the study samples. Lastly, they used ALDEx2 to examine the differential abundance of microbial taxa across the four study groups.
Study findings
The longer study duration provided access to clinical specimens positive for a range of SARS-CoV-2 variants. While all the study participants had similar age distribution, there were 60.3% males and only 39.7% females. The authors observed a trend towards lower mean Shannon/Simpson diversity and evenness in samples from COMPOS vs. COMNEG groups. However, there was no significant alpha diversity among all the study groups, as indicated by Kruskal-Wallis test results with p-values less than 0.05. Further, the authors observed a degree of overlap in the 95% confidence ellipses among four study groups; however, they differed in their microbial community composition with significant clustering at all three taxonomic ranks, species-, genus-, and family level. Accordingly, p-values for species-level, genus-level, and family-level taxonomic ranks were 0.009, 0.028, and 0.027, respectively.
In the hospitalized and the community-dwelling SARS-CoV-2-infected groups, Acinetobacter and Moraxella were the most abundant genera, respectively. In both SARS-CoV-2-uninfected groups, Staphylococcus was the most abundant genus on average. At the species level, the SARS-CoV-2 infected groups had common nasal and opportunistic pathogens, including Haemophilus influenzae, Staphylococcus haemolyticus, and Staphylococcus aureus. Notably, the SARS-CoV-2-infected hospitalized group had Klebsiella aerogenes as the most abundant species and a higher mean relative abundance of Enterobacteriaceae than any other study group.
The community-dwelling SARS-CoV-2 infected individuals exhibited all the differentially abundant taxa, such as the families Propionibacteriaceae, Neisseriaceae, Peptostreptococcales-Tissierellales, the species Cutibacterium acnes, and the genera Cutibacterium and Peptinophilus.
ALDEx2 revealed significant differences in Streptococcus. and Corynebacterium species among samples from SARS-CoV-2-infected individuals collected during the first three waves of the COVID-19 pandemic between March 2020 and May 2021. However, the authors did not observe any significant alpha or beta diversity linked to viral load or by SARS-CoV-2 VOC type.
Conclusions
Overall, the study results presented a granular picture of the microbiota differences in SARS-CoV-2-infected and uninfected individuals. Despite significant overlap in the confidence ellipses, the differences in nasopharyngeal microbiota composition among study groups were relatively small.
A non-influenza respiratory viral infection increases the host’s susceptibility to S. aureus superinfection by reducing the immune system’s ability to regulate its clearance from the nasal passage. Therefore, the medical interventions such as intubation or antibiotic exposure most likely increased the abundance of Enterobacteriaceae among the hospitalized SARS-CoV-2-infected study group. However, the presence of Neisseriaceae taxa in the community-dwelling SARS-CoV-2-infected group remains unexplained.
Further work is needed to determine the functional characteristics of the nasopharyngeal microbiome. This knowledge could help develop novel COVID-19 prognostic markers and open new avenues for new approaches to COVID-19 treatments.
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Alterations in the Nasopharyngeal Microbiome Associated with SARS-CoV-2 Infection Status and Disease Severity, Nick P.G Gauthier, Kerstin Locher, Clayton MacDonald, Samuel D Chorlton, Marthe Charles, Amee R. Manges, medRxiv pre-print 2022, DOI: https://doi.org/10.1101/2022.06.13.22276358, https://www.medrxiv.org/content/10.1101/2022.06.09.22276196v1
- Peer reviewed and published scientific report.
Gauthier, Nick P. G., Kerstin Locher, Clayton MacDonald, Samuel D. Chorlton, Marthe Charles, and Amee R. Manges. 2022. “Alterations in the Nasopharyngeal Microbiome Associated with SARS-CoV-2 Infection Status and Disease Severity.” Edited by Sylvia Maria Bruisten. PLOS ONE 17 (10): e0275815. https://doi.org/10.1371/journal.pone.0275815. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0275815.
 Can an allergy spray help prevent COVID-19?
Can an allergy spray help prevent COVID-19?