Microbial communities play a crucial role in the human body, influencing the immune system and anticancer therapies. They are present in primary tumors and interact with the commensal microbiota. The gut microbiota can modulate immune checkpoint blockers (ICB) and conventional chemotherapies. Fecal microbial transplants may improve clinical responsiveness to ICB agents. Understanding how tumor-resident bacteria shape tumor biology, immune infiltration, and treatment responsiveness is essential for understanding tumor response to ICB.
 Study: A pan-cancer analysis of the microbiome in metastatic cancer
Study: A pan-cancer analysis of the microbiome in metastatic cancer
About the study
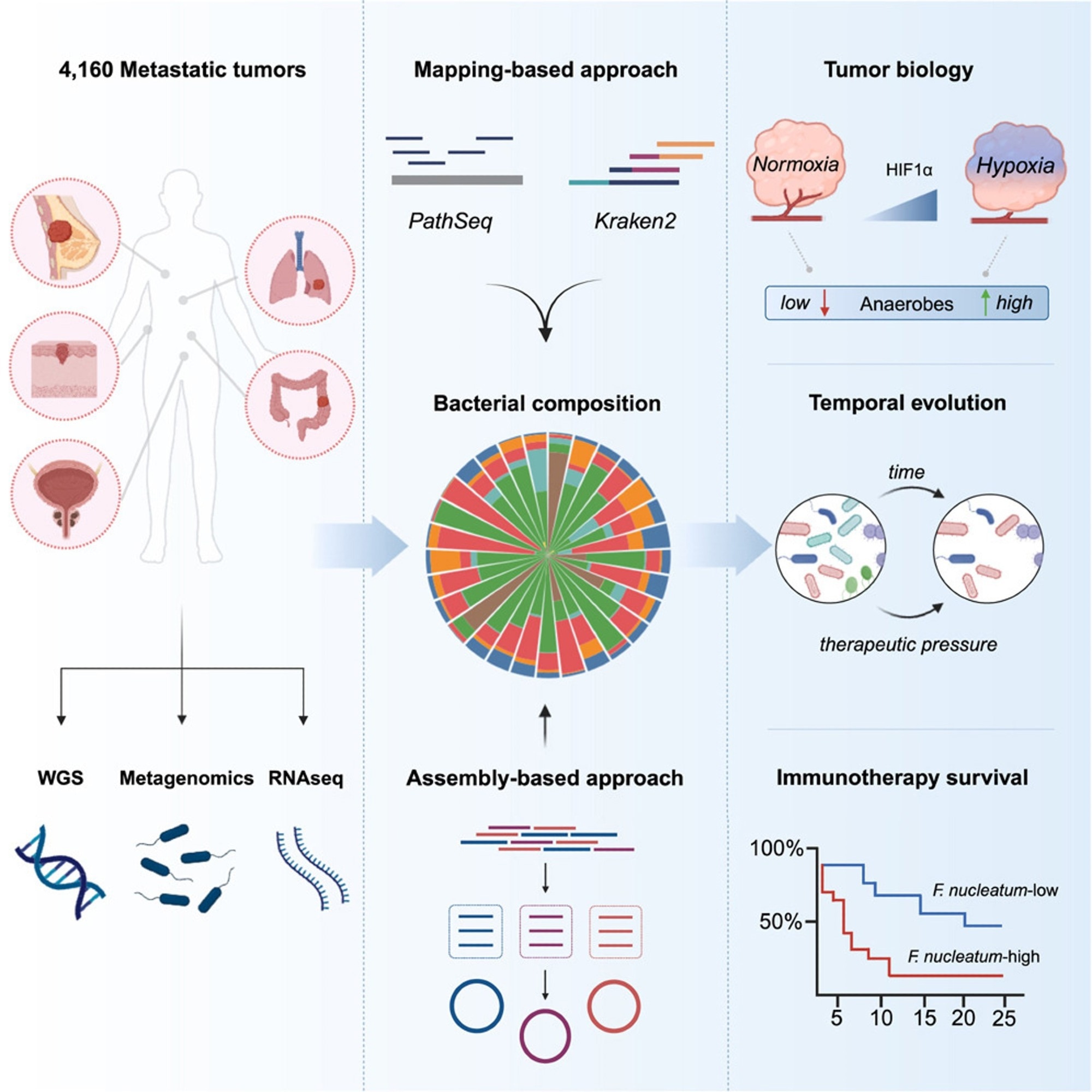
In the present study, researchers used bioinformatics to investigate the microbiota in metastatic malignancies, evaluating 4,160 specimens from diverse cancer types.
The researchers used mapping and assembly-based metagenomics, genomes, transcriptomics, and clinical data to develop a pan-cancer repository that might help advance treatment techniques. They used two distinct computational approaches, PathSeq and Kraken2, to define tumor-resident microbiome communities at the genus level and a metagenomic assembly-based approach at the species level. The team then shaped the metastatic tumor microbiome by identifying the elements that influence its makeup and evaluating cancer-type-specific microbial communities. They used the characteristic hypoxia gene profile to assess the degree of hypoxia in metastatic cancers and then performed gene set enrichment analysis (GSEA). They also investigated whether microbial communities may affect host immunity and the TME.
The researchers investigated the relationship between gram-negative bacteria in metastases and Toll-like receptor (TLR) expression and whether lipopolysaccharide (LPS), obtained from dead or active bacteria, plays a primary role in TLR4 signaling in metastases. They additionally examined the relationship between bacterial makeup and tumor gene expression and the relationships between particular bacteria and immune cells.
To further understand the impact of metastatic heterogeneity and the durability of tumor-resident microorganisms over time, the team examined 185 pairs of 370 repeated tumor specimens obtained from 173 different individuals. They examined bacterial enrichment changes before and after tumor treatment with immunotherapy, targeted therapy, or hormone therapy. They also investigated bacterial count reductions following immunotherapy in responsive patients and whether these germs were more prevalent in non-responsive individuals before treatment. Lastly, they examined pre-treatment bacterial communities associated with a lack of response to immunosuppressive medication in an ICB-monotherapy cohort of NSCLC patients.
Results
The researchers detected tumor-resident bacteria deoxyribonucleic acid (DNA) in a pan-cancer metastasis cohort, and assembling tumor-derived bacterial DNA provided species-level genomic characterization. Bacterial diversity correlated with cellular and molecular tumor immunity characteristics. In an NSCLC cohort, high levels of fusobacterium DNA imply a poor immunotherapy response. Researchers found organ-specific microbe tropisms, anaerobic bacteria enrichments in hypoxic tumors, links between microbial diversity and tumor-infiltrating neutrophils, and Fusobacterium's relationship with resistance to ICB therapy in lung cancer.
Using mapping-based techniques and screening genera to eliminate technical contamination and seldom-seen genera, the team cataloged 165 microbial genera from 3,526 specimens, with 68% facultative/anaerobic and 49% gram-negative anaerobes. They built 514 metagenomic-assembled genomes (MAGs) of medium- to almost high-quality using tumor-derived microbial sequences. The most common tumor types were colorectal, breast, prostate, lung, and melanoma, with the lymph node, liver, and lung being the most common metastatic locations for tumor samples.
The quantity of bacterial-derived reads expressed as a human-mapped genetic read proportion varied by cancer type, with higher fractions in renal and uterine malignancies and lower burdens in tumors originating from the brain and spinal cord. Renal and colorectal metastases were the most diverse, but head and neck metastatic tumors showed more dominant microbial genera.
Tumor-resident microbial communities were associated with tumor biology, with a strong correlation between LPS load and TLR4 signaling but not gram-positive lipoteichoic acid (LTA) load. Multivariate Cox proportional-hazards modeling showed lower overall survival (OS) and progression-free survival (PFS) rates significantly correlated with continuous Fusobacterium abundance, considering the genome-wide mutational load. Using the pan-cancer dataset, the researchers classified all tumors as Fuso-high or Fuso-low based on an upper quartile relative abundance cutoff similar to previously established criteria. Fuso-high tumors showed considerably decreased cytotoxic, interferon-gamma (IFN-γ), and major histocompatibility complex (MHC) class II gene expression profiles.
The study provides the first large-scale pan-cancer map of intratumor microbiomes in metastatic malignancies, examining diversity across anatomical regions, initial tumor type, and treatment responses, including immunotherapy. The study showed that the metastatic microbiome partially comprises anaerobic bacteria that may get altered during treatment. The study also discovered links between intra-tumoral microorganisms and the activation of innate immune sensing pathways, indicating that the tumor microenvironment alters via direct identification of bacterial ligands.
 Dual targeting strategy suppresses pancreatic cancer cell growth
Dual targeting strategy suppresses pancreatic cancer cell growth