This article provides a summary of Biognosys’ Spectronaut 15 Launch Seminar, featuring lead developer Oliver Bernhardt from Biognosys and Andreas-David Brunner from Matthias Mann’s lab at the Max Planck Institute of Biochemistry.
The primary goal of proteomics is the comprehensive identification and quantification of the entire set of all expressed proteins in a cell, tissue, or organism. The study of proteins provides insights into the functioning of organisms, enabling breakthrough discoveries that bring life science to a new level. Mass spectrometry-based proteomics is the leading technology to comprehensively and accurately measure proteins and gain multi-dimensional insights into the proteome at large scale.
There are two main ways of using mass spectrometry to acquire large-scale protein information: the traditional data-dependent acquisition (DDA) method, and the next-generation data-independent acquisition (DIA) method. In DDA, individual peptide ions are selected as precursors for fragmentation in a semi-stochastic manner, favoring the most intense peaks and losing the rest. As a result, a very low percentage of the detectable peptides get fragmented, and an even lower number is reliably identified.
In DIA, in contrast, all precursors are fragmented, and data is acquired for all fragment ions. This allows recording MS1 and MS2 data from virtually all peptides in a sample without the loss of information while enabling highly accurate quantification at MS2 level. MS2 data is more complex, however, requiring the use of specialized software (such as Spectronaut) to deconvolute MS2 spectra and perform quantification.
Leveraging directDIA™ for Deep Proteome Coverage
Traditionally, DIA data analysis requires a spectral library that is still based on DDA data. In contrast, Biognosys' proprietary directDIA workflow can analyze DIA data without an additional library. directDIA is a two-step process: first, DIA data is extracted into a collection of MS2 spectra, which are searched using Spectronaut's integrated Pulsar search engine. Next, search results are used to generate a spectral library, which is employed in the targeted analysis of DIA data.
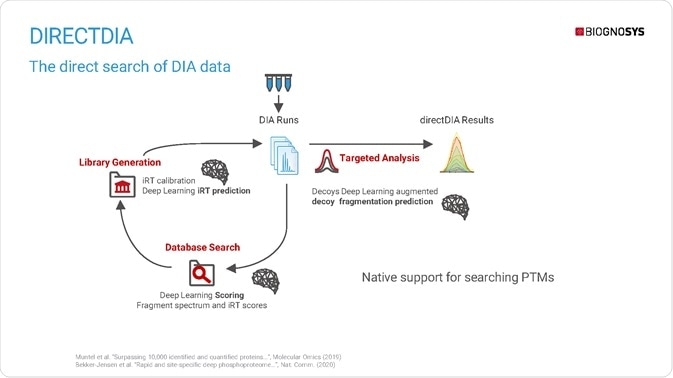
directDIA natively supports the searching of post-translational modifications, while deep learning predicts peptides' physical and chemical properties at different stages of the directDIA pipeline for fragmentation and retention time prediction.
directDIA has the potential to streamline proteomics while achieving comparable proteome coverage to project-specific DDA libraries – it is even possible to break the 10,000 protein identification barrier when directDIA is combined with state-of-the-art instrumentation.
Introducing Spectronaut 15
Spectronaut 15 boasts a diverse array of additions and improvements over previous versions of the software. These include improvements to general identification performance in classic library-based DIA analysis as well as directDIA, particularly for dia‑PASEF data.
General improvements have also been made to quantification strategies, the post-translational modification (PTM) analysis workflow, the user interface, and the software's false discovery rate (FDR) control; including additional flexibility to set strict criteria and apply run-level protein FDR and peptide-level posterior error probability (PEP) thresholds.
As mentioned above, Spectronaut 15 incorporates a new and improved PTM workflow, offering sophisticated post-processing and site-level summarization, often required to interpret PTM data properly. This PTM workflow features a PTM site report including a number of key aspects; for example, the flanking region around the modified amino acid, the modification site location within the parent protein, and PTM site quantity - useful for downstream analysis. An improved protein coverage plot is also available, alongside a post-analysis process that enables regulation analysis directly on PTM sites.
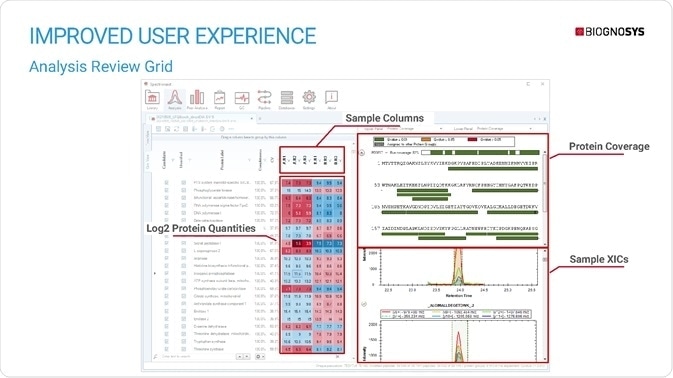
Spectronaut 15 also features a new way of browsing and visualizing data. The grid view allows users to easily see protein-level information across runs, including protein quantities. Detachable perspectives and floating plot windows allow users to explore several views of the data in parallel. Users who routinely work with dia-PASEF data will benefit from the addition of the ion mobilogram plot – a novel option for data browsing, including peak integration.
Deploying Spectronaut 15 for Large-Scale Single-Cell Proteomics
Single-cell proteomics can provide unique insights into cellular processes in disease and health with protein expression data for individual cells. Achieving high coverage of the single-cell proteome is particularly challenging and requires highly sensitive instrumentation as well as a state-of-the-art data analysis pipeline.
Andreas-David Brunner and his colleagues from Matthias Mann’s lab at the Max Planck Institute of Biochemistry have recently developed a pioneering DIA single-cell proteomics workflow that includes Spectronaut as the solution for data analysis.
The team used a modified version of their Bruker timsTOF Pro (?) mass spectrometer alongside a novel liquid chromatography system to capitalize on the high sensitivity afforded by ion mobility mass spectrometry. From only one nanogram of total digested proteins, they identified over 3,500 protein groups using Biognosys’ Spectronaut 15, almost 10% more identifications than with the previous version.
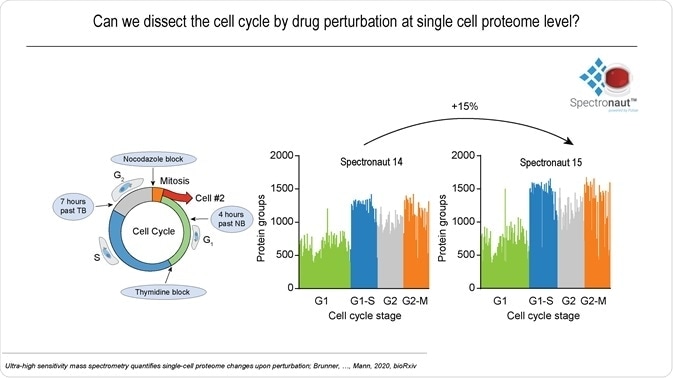
A large-scale single-cell experiment was conducted on whether it was possible to dissect cell cycle-dependent proteome changes using drug perturbation. Single cells at four cycle stages were investigated using the developed LCMS setup and Spectronaut 15, identifying over 1,500 proteins in many individual cells, on average 15% more than with the previous version.
Correlating transcriptomics and proteomics data further showed that both offered different yet complementary information. Taken together, these results demonstrate that a combination of the latest acquisition methods and software innovations are driving DIA-based single-cell proteomics towards a prominent role in clinical and biomedical decision-making. Learn more about how Spectronaut 15 can advance your proteomics projects today!
About Biognosys
Biognosys is the leading proteomics company offering innovative services and products for highly multiplexed protein quantification. Founded in 2008 as a spin-off from the lab of proteomics pioneer Ruedi Aebersold at ETH Zurich, Biognosys is dedicated to transforming the life sciences with next-generation proteomics solutions.
Biognosys believes that the decoding of the proteome will impact the life sciences more than the genome revolution two decades ago. Biognosys’ next-generation technology quantifies proteins with unbeatable precision and depth. Our solution relies on mass spectrometry, which allows simultaneous quantification of thousands of proteins in a single experiment. This new generation protein quantification technology is available to researchers worldwide through our contract research services or our portfolio of innovative reagent and software products.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.