The coronavirus disease 2019 (COVID-19) pandemic is caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is part of a family of viruses characterized by the presence of spike proteins on their surface envelopes. The spike is not a decorative accessory but a crucial component of the virus, mediating its entry into the host cell.
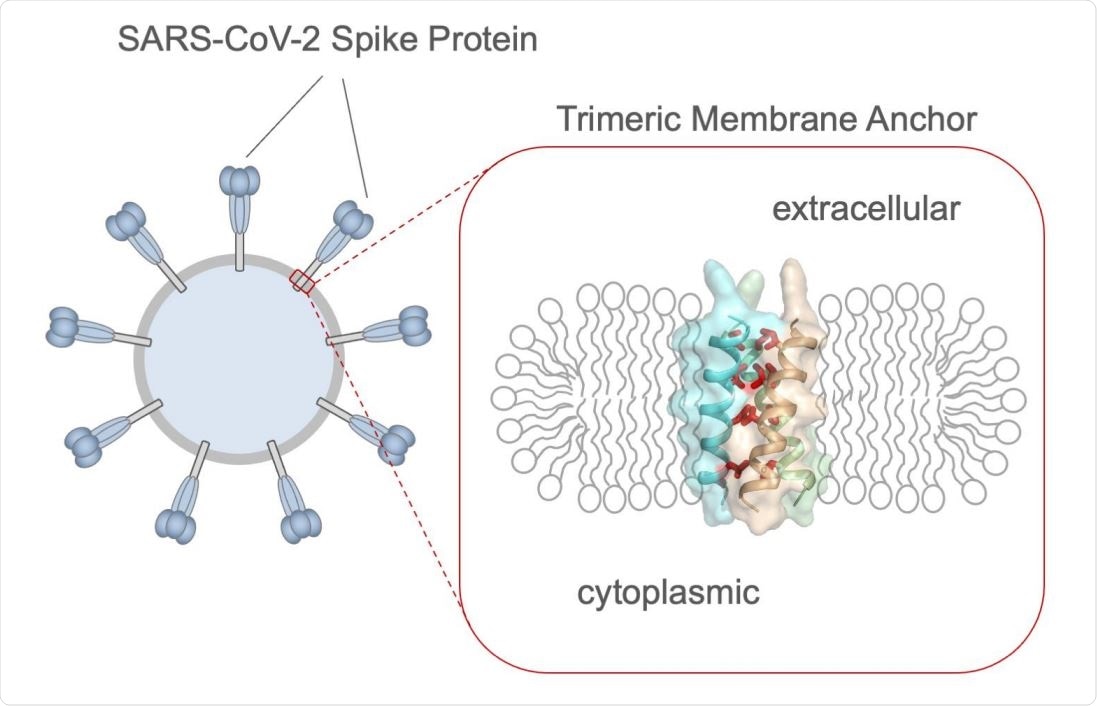
The spike TMD
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
SARS-CoV-2s spike protein is the main viral target of the host immune system, with the crown-shaped part being the ectodomain. This is composed of the receptor-binding domain (RBD), the transmembrane domain (TMD), and a cytoplasmic tail (CT). The NMR focused on the TMD, using a bicellar structure to mimic a lipid bilayer.
The TMD has important functional activity, rather than being a simple anchoring protein. When the SARS-CoV-2 spike is substituted with the vesicular stomatitis virus (VSV) G protein, the resultant particle had 3% to 25% spike activity relative to the wildtype virus.
In an earlier study, an insertion point mutation in the TMD abolished viral entry. This protein also appears capable of trimer formation, as shown on directly fusing the RBD to the TMD.
The current study shows that the spike TMD forms a trimer using a transmembrane helix mode of assembly, seen in this context for the first time.
 Ribbon representation (left) of the TMH trimer structure with the sidechain heavy atoms of the core residues shown as spheres; the C atoms of G1219 and G1223 are also shown as spheres. The sidechain packing at four different levels along the threefold axis is illustrated with sectional top views of the trimer (right). (b) Helical wheel representation of an -helix (3.6 residues per turn) showing that the core hydrophobic residues occupy the position ‘a’ of the ‘abcd’ tetrad repeat. (c) Theoretical analysis of the trimeric hydrophobic zipper with tetrad repeat. The line formed by the C atoms of the residues at position ‘a’ is tilted by ~17º relative to the helical axis. Rotating the helix by 17º places the position-a residues in line with the 3-fold axis for optimal hydrophobic core formation.")
NMR structure of the TMH trimer of SARS-CoV-2 S in DMPC-DH6PC bicelle with q = 0.55. (a) Ribbon representation (left) of the TMH trimer structure with the sidechain heavy atoms of the core residues shown as spheres; the C atoms of G1219 and G1223 are also shown as spheres. The sidechain packing at four different levels along the threefold axis is illustrated with sectional top views of the trimer (right). (b) Helical wheel representation of an α-helix (3.6 residues per turn) showing that the core hydrophobic residues occupy the position ‘a’ of the ‘abcd’ tetrad repeat. (c) Theoretical analysis of the trimeric hydrophobic zipper with tetrad repeat. The line formed by the Cα atoms of the residues at position ‘a’ is tilted by ~17º relative to the helical axis. Rotating the helix by 17º places the position-a residues in line with the 3-fold axis for optimal hydrophobic core formation.
Tetrad repeats stabilize the TMH helix
The S2 subunit showed a short stretch of the membrane-proximal region and the TMD, reconstituted as bicelles, when it formed trimers. The first stage was the formation of a TMD alpha-helix, which then assembled to form a homotrimer with three parallel helices.
The NMR visualization showed a hydrophobic core holding the trimer together along its three-fold axis, with four layers of hydrophobic interaction between four residues. This core contains a tetrad repeat pattern, with I1221, I1225, L1229, and L1233 being seen at position 1 of a four-member repeat.
The tetrad repeats form a novel hydrophobic zipper motif. More typically, TMH assembly uses a coiled-coil mode involving heptad repeats of isoleucine and leucine. The tetrad repeat pattern, in this case, means that the repeat is longer than a single turn of the helix, which is 3.6 residues long. This results in a 400 extension past the end of the turn, shifting the hydrophobic core ridge from the three-fold axis by 170 each time.
This allows the three hydrophobic ridges of the trimeric TMHs to fall in line with the three-fold axis, and thus packs them closely, forming a stable structure.
Novel mode of assembly
Interestingly, glycine and alanine sequences are more typically involved in the assembly of transmembrane helices (TMH), being small amino acids. While these are certainly present in this structure, they do not take part in forming close van der Waals interactions.
Instead, the four bulky lysine residues are part of the hydrophobic core. Thus, this mode of TMH trimer formation differs from the conventional mode.
“The trimeric complex is stabilized by an extensive hydrophobic core along the 3-fold axis, formed by the bulky hydrophobic amino acids repeated every four residues.”
This structure appears to be stable in the membrane, which may imply that “the TMD trimer is unlikely to dissociate in the membrane unless significant force is applied during the unfolding and refolding steps of the fusion component.”
Effect of designer mutations
The researchers also looked at what would happen if they inserted seven point mutations involving glycine, alanine, and lysine residues in the TMD region. They found that changing the glycine/alanine sequence to tyrosine did not affect the formation of TMH trimers.
Both of the glycine residues here face the lipid face of the membrane, while the alanine residue is not positioned deep enough, and should not, therefore, take part in helix-helix van der Waals packing.
Conversely, lysine mutations here interrupt the process of trimerization. This is especially true of the more central mutations, which almost abolish the process, while the more peripheral ones partially disrupt it.
The trimeric TMH is also very stable, with no hydrogen-deuterium exchange occurring over weeks.
What are the implications?
The study describes a highly stable novel zipper motif that achieves trimeric assembly of the TMHs. The findings underline the importance of the tetrad repeat of large hydrophobic residues in this process. The study also identified point mutations capable of dissociating the trimeric TMH.
While it has been known for some time that the membrane anchor domains of viral fusion proteins do form specific oligomers, the role of such oligomers has remained unclear. Further development of the current results could help understand how they are involved in the function of viral fusion proteins, in general.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
 Scientists discover spindle's self-repair mechanism for accurate cell division
Scientists discover spindle's self-repair mechanism for accurate cell division