Two years after it was first detected in Wuhan, China, severe acute respiratory syndrome coronavirus 2 (SARS‑CoV‑2) has caused over 282 million infections worldwide. SARS-CoV-2 is a betacoronavirus that arose by zoonotic transmission from an animal reservoir, most likely the horseshoe bat (Rhinolophus), through an intermediate host. As the virus has spread throughout society, it has evolved and adapted to maximize its fitness in the human population. Consequently, viral variants that are more transmissible and resistant to therapeutics and vaccine-induced antibodies are emerging.
Most new variants of SARS-CoV-2 result from mutations in its spike protein. A homotrimeric transmembrane protein, the spike protein is essential for the pathogenicity of the virus. It is split into two subunits that require a host protein to cleave them for activation, S1, and S2. S1 contains a receptor-binding domain (RBD) that can bind to angiotensin-converting enzyme 2 (ACE2), alongside other receptors, in order to permit viral cell entry, while S2 mediates membrane fusion.
Now in new research, scientists from the University of Leicester have been investigating how these mutations alter the affinities of RBDs to ACE2. The groups' study can be found on the bioRxiv* preprint server.
The Study
The researchers used the DT40 cell surface display system to identify spike variants that could potentially show an increased affinity for ACE2. DT40 cells were transfected to produce a fusion protein with the secretory leader sequence from CD5, the RBD domain, a linker region, and a FLAG-epitope tag, followed by the transmembrane domain and short intracellular fragment from platelet-derived growth factor receptor-beta. These cells were selected for antibiotic resistance, and ACE2 binding was determined by incubating cells with monomeric ACE2 residues with a tag and bound ACE2 detected with anti-histidine, while RBD expression was detected using anti-FLAG. Diagonal sort windows were used to normalize for RBD expression. Rounds of selection were limited to three to favor variants with a low number of accessible mutations, and sort windows were used to capture variants that showed the most significant gain in affinity. The dominant variant that the scientists found was designated RBD4.8 and showed double substitutions S477N and Q498H. These mutations have already been found in variants in patients, and have emerged independently several times – although not within the same strain.
The scientists used biolayer interferometry to measure the binding ability of wild-type RBD and mutant RBDs with ACE2. Wild-type RBD was found to bind with a Kd of 21.2nm, whereas the RBD4.8 shows a significant increase in binding affinity, around 1.1Kd. This is representative of a 19-fold increase in binding affinity. When assessing the two mutations individually, RBD carrying the S477N mutation showed a Kd of 4.3nM, while Q498H showed a Kd of 2.0nm, one of the highest increases in binding affinity from a single substitution that has been observed. Flow cytometry was used to confirm that this increase is reflected in a cellular context, with Vero-E6 cells expressing ACE2 incubated with the different RBDs, displaying similar results to the biolayer interferometry.
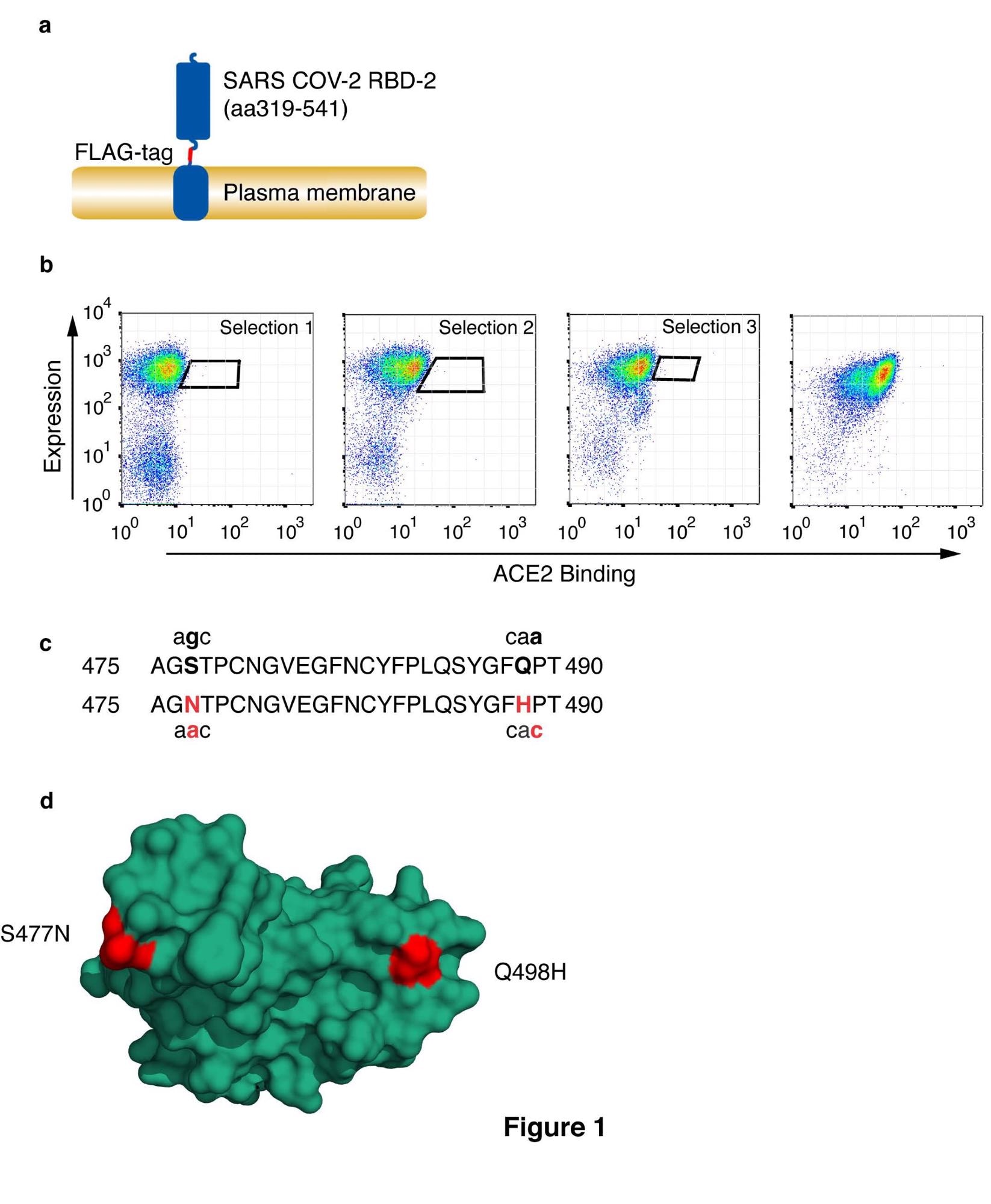
Selection of CoV-2 receptor binding domain with high affinity for HuACE2. (a) Schematic representation of cell surface-expressed CoV-2 RBD showing RBD, FLAG-epitope tag, and PDGF-receptor transmembrane anchor. (b) CoV-2 RBD was selected for enhanced ACE2 binding by somatic hypermutation and cell surface display in DT40 cells. FACS plots are shown of DT40 cells following binding of H6-tagged-HuACE2. Bound ACE2 was detected with anti-Histidine6-PE and the expression level of RBD was assessed with ant-FLAG-APC. Sort windows are indicated for each round of selection and diversification, selecting at each round for the highest Ang2 binding. Selections were performed at 0.1nM ACE2. The final panel shows a binding plot of the selected cells. (c) Amino acid substitutions S477N and Q498H in the selected RBD are shown, along with the corresponding single-nucleotide mutations. Substituted residues and nucleotides are in red. (d) Position of residue substitutions in the selected RBD. Substituted residues are shown in red on the ACE2 binding interface (PDB accession number 6M0J, 20). The RBD is orientated to show the face that interacts directly with ACE2.
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Following this, the researchers decided to investigate the structure of the RBD4.8:ACE2 complex in order to understand the binding mechanism better. Cryo-EM provided a map upon which an atomic model for the complex could be built. This model revealed that substitution of RBD Q498 for histidine results in a perpendicular Y-shaped interaction between ACE2 Y41 and RBD H498. The RBD residues also now lie within the hydrogen bonding range of another ACE2 lysine, further strengthening the interaction. ACE2 D38 can also mediate a hydrogen bond via a water molecule to RBD H498, supported by a continuous density. A newly gained carbon atom places another side chain in hydrogen bonding distance on the other side of the interface. Further examination of the differences between the new binding interface reveals that the RBD Q498 hydrogen bonding changes. The positive difference in electron density adjacent to the side chain is consistent with disorder.
The researchers added the Q498H substitution to two prominent variants, containing the substitutions L425R and E484Q or K417N, E484K, and N5017 – the Kappa and Beta variants. This allowed them to test whether this substation could enhance binding in other variants. In Beta, the substitution actually reduced the affinity to ACE2, but the Kappa variant showed significant increases in affinity.
Structure of RBD4.8 in complex with HuACE2 Cryo-EM structure and model of the ACE2-RBD complex. Sharpened Cryo-EM map (a) of the HuACE2-RBD4.8 complex with ACE2 colored yellow and RBD in green. The refined coordinates are shown in cartoon representation (b) and colored as above. In panel (c) H498 can be seen in proximity to ACE2 Y41 forming a non-planar π-interaction while ACE2 residues K353 and D38 are within hydrogen-bonding distance to H498 and could contribute to the tighter interaction formed by this RBD mutant, whereas Panel (d) shows Q498 in RBD1 (PDB accession number 6M0J, 19) in proximity to ACE-2 Q42. Panel (e) indicates that the S477N substitution places this longer side-chain closer to S19 in ACE2 and within hydrogen-bonding distance thus enhancing the binding between HuACE2 and RBD4.8. Panel (f) shows the positioning of S477 in RBD1 and ACE2 S19 (PDB accession number 6M0J19). (g) Biolayer interferometry was performed with RBD containing the Q498R substitution in solution and HuACE2 immobilized on the sensor. Curves were fitted and used to calculate Kon, Koff and KD.
The reduction in affinity seen in Beta is likely due to the constraining of the local arrangement required to accommodate the phenolic group of Y501 by the imidazole of H498. This will reduce the pi-pi stacking between Y501 and Y41 as well as perturbing the H498-Y41 interactions. However, in the Kappa variant, the arginine can be accommodated at 498 in an N501Y carrying variant, resulting in a large affinity gain by a bonding mechanism that requires R498 as well as Y501, sharing similarities with variants that carry H498 on its own.
Conclusion
The authors highlight that they have shown how the different mutations and substitutions change the binding affinity of the SARS-CoV-2 spike protein RBD to ACE2, not only in isolation but when introduced in prominent variants of concern. Further examination of the structure helps to reveal the mechanics behind this. This information could be invaluable in informing epidemiologists and could help sway public health policy in the future.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
In vitro evolution predicts emerging CoV-2 mutations with high affinity for ACE2 and cross-species binding. Neil Bate, Christos G Savva, Peter CE Moody, Edward A Brown, Jonathan K Ball, John WR Schwabe, Julian Sale, Nicholas Brindle. bioRxiv 2021.12.23.473975; doi: https://doi.org/10.1101/2021.12.23.473975, https://www.biorxiv.org/content/10.1101/2021.12.23.473975v1
- Peer reviewed and published scientific report.
Bate, Neil, Christos G. Savva, Peter C. E. Moody, Edward A. Brown, Sian E. Evans, Jonathan K. Ball, John W. R. Schwabe, Julian E. Sale, and Nicholas P. J. Brindle. 2022. “In Vitro Evolution Predicts Emerging SARS-CoV-2 Mutations with High Affinity for ACE2 and Cross-Species Binding.” Edited by Tyler Starr. PLOS Pathogens 18 (7): e1010733. https://doi.org/10.1371/journal.ppat.1010733. https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1010733.
 Poor lung gas exchange may help explain lingering brain symptoms in Long COVID
Poor lung gas exchange may help explain lingering brain symptoms in Long COVID