The coronavirus disease 2019 (COVID-19) pandemic has significantly impacted all nations across the globe, causing unprecedented morbidity and mortality. Since the release of the first SARS-COV-2 genetic sequence on January 5, 2020, the deleterious effects of its genetic clades due to the presence of several mutations have been reported.
Understanding the genetic aspects and regional spread of SARS-COV-2 could aid in the development of more efficient and targeted vaccines. Thus, the researchers of the present study investigated the temporal occurrence and geographical localization of the multiple mutated SARS-COV-2 variants.
About the study
The study explored The National Center for Biotechnology Information (NCBI) and the Global Initiative on Sharing All Influenza Data (GISAID) databases between December 2019 and September 2020, from which 77,648 viral genomes were identified.
Data including their geographical distribution, sampling date, and length of the genetic sequences were obtained. Only 75,401 genomes with sequences exceeding 29,000 nucleotides were analyzed. Fifty-three variants present in greater than 1,000 genetic sequences were classified into clades, and their haplotypes were counted. Only nations with variants exceeding 50 sequences were chosen for the analysis.
Results and discussion
Using the earliest SARS-CoV-2 sequence identified in Wuhan as a reference, the authors observed 26,539 mutations of several types such as missense (57%), synonymous (28%), insertions/deletions (7%), and stop variants (2%), from which 4% and 3% were present in the 3’and 5’ untranslated regions, (UTR) giving rise to five to nine mutant variants. These variants with similar geographical spread were grouped into four genetic clades.
A majority (58%) of the sequences were obtained from Europe, especially from the United Kingdom, followed by Oceania, North America, and the least in Asia. Most mutated variants were observed in France and Italy, whereas most unmutated variants were present in China, followed by the USA, Northern Europe, Australia, South Africa, Brazil, Canada, and India.
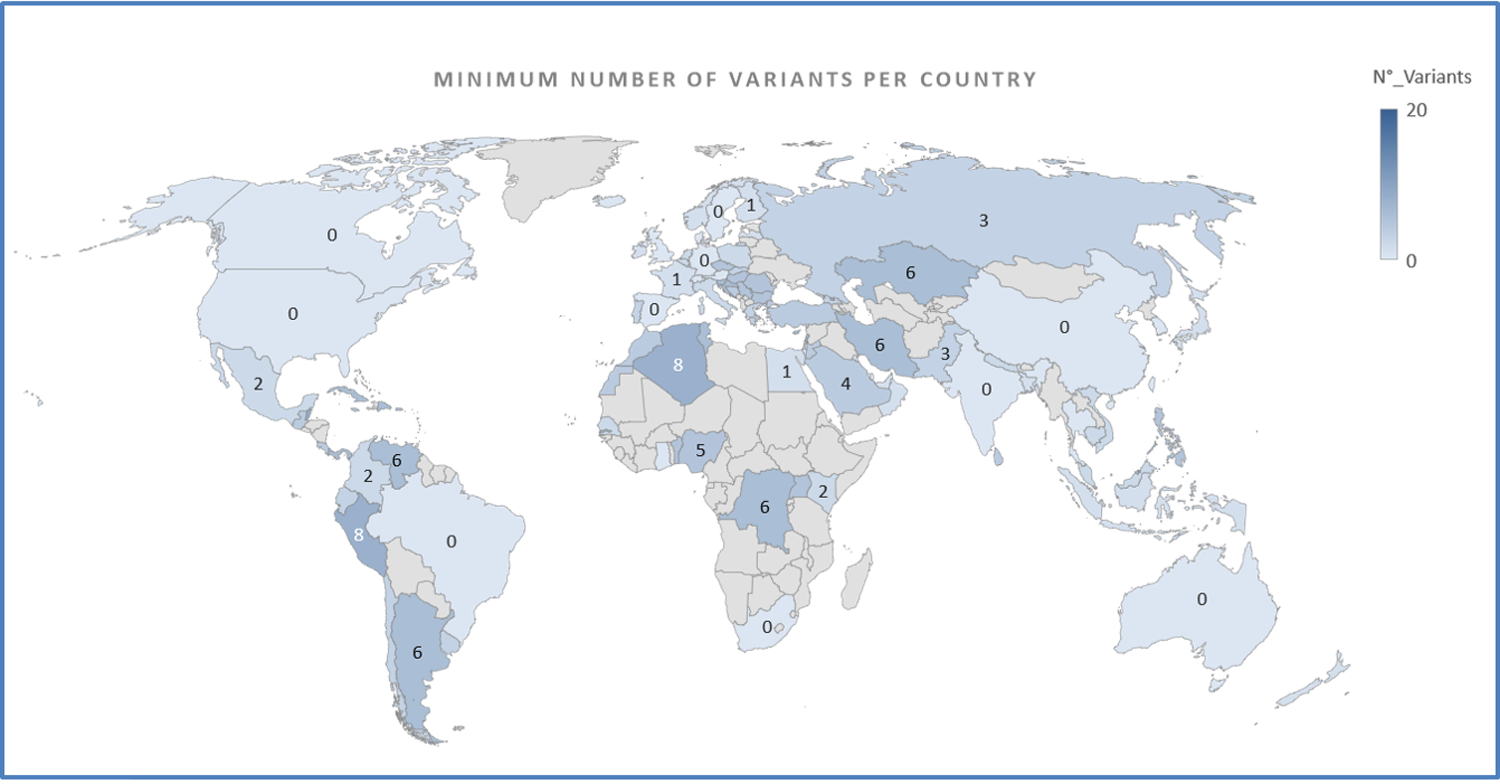 Geographical distribution of the minimum number of variants per sequence. https://doi.org/10.1371/journal.pone.0263705.g001
Geographical distribution of the minimum number of variants per sequence. https://doi.org/10.1371/journal.pone.0263705.g001
The most frequently reported and highly infectious p.Asp614Gly mutant variant with a missense mutation in its S glycoprotein was observed in combination with other three variants: the p. Pro4715Leu, the c.-25C>T, and the p.Phe924Phe situated in the 5’ untranslated region (UTR) on the ORF1ab gene. These variants were grouped into the first clade, observed in 55,582 (74%) genetic sequences.
Reported earliest by the UK in European countries in January 2020, they were also observed in Italy (93%), northern (73%) and southern parts (77%) of America, Oceania (73%), Africa (77%). This genetic clade produced two subclades, IA and IB observed majorly in Oceania and North America, respectively.
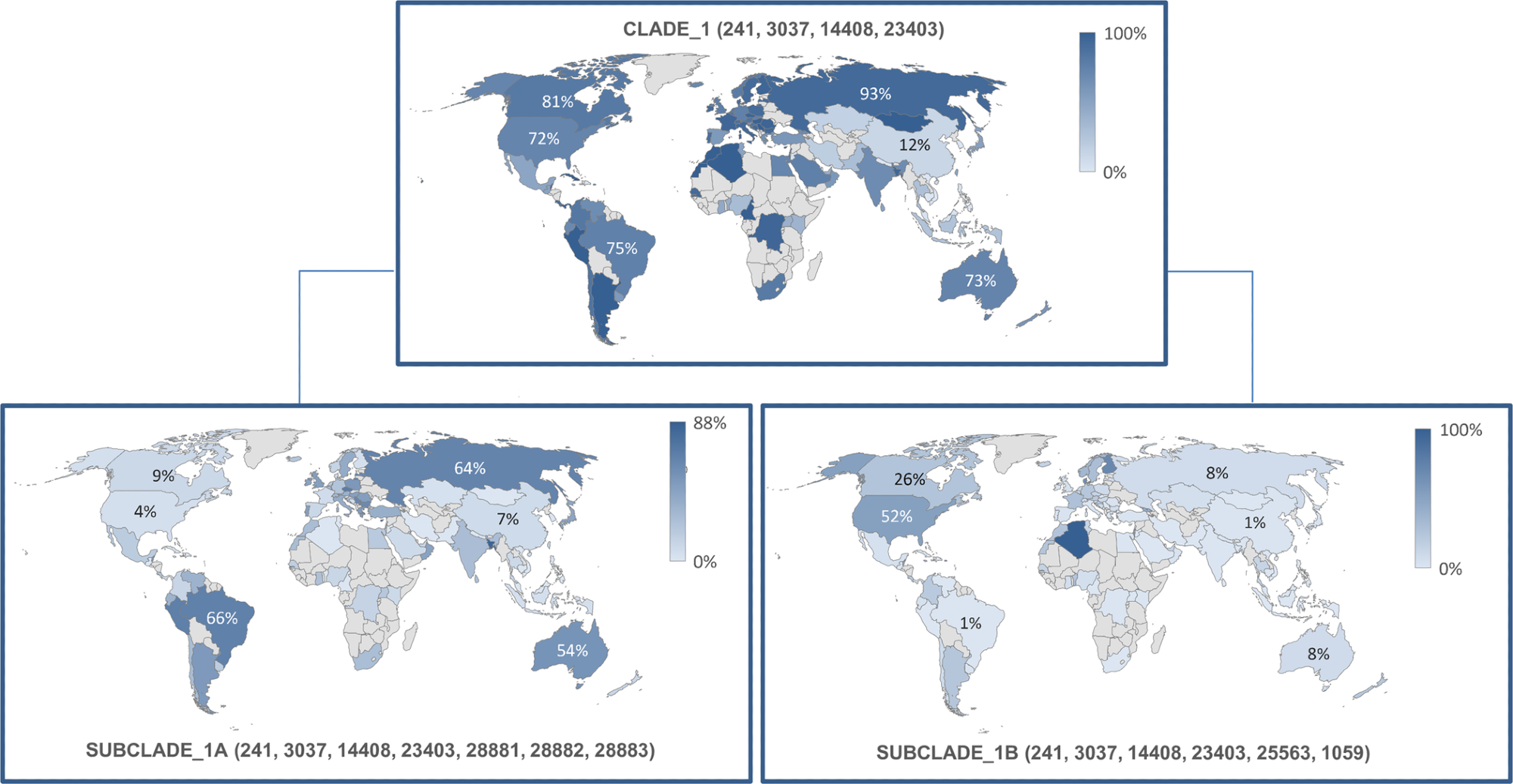 Frequency and geographical distribution of sub-clades 1A and B. https://doi.org/10.1371/journal.pone.0263705.g002
Frequency and geographical distribution of sub-clades 1A and B. https://doi.org/10.1371/journal.pone.0263705.g002
The second clade comprising two variants was present in North America, Spain, Asia (China, Thailand, and Kazakistan), Australia, and Africa (Nigeria and Ghana). The third and fourth clades were majorly observed in Europe. Singaporean and Australian genetic sequences comprised of five and six variants, respectively, whereas Kazakistan and Spain demonstrated the presence of four variants in their genetic sequences.
The four genetic clades constituted the center of 1213 haplotypes. The first, second, and third haplotypes were most commonly observed in South America and Europe, mainly spreading to North America, Africa, and Europe.
Several genetic variants were localized to specific countries, for example, the 313 genetic variants localized in Japan and the variants 29829 and 18877 restricted to Saudi Arabia. The p.Asn501Tyr genetic variant was observed in USA and Brazil in early April and had spread to Australia by June 2020.
Conclusion
Based on the present study results, the authors concluded that multiple SARS-CoV-2 mutated variants exist globally, majorly grouped into four clades with specific geographical localization, representative of the spatial and temporal spread of the mutant strains.
Another observation pointed out by the researchers was that synergistic and simultaneous action of genetic mutations could confer significant clinical advantages to the viral variants in terms of high viral transmission rates and infectivity by enhanced binding to the angiotensinogen conversion enzyme 2 (ACE2) receptors on human cells.
The study findings highlight the stronghold of the highly infective SARS-CoV-2 virus across the globe. Therefore, strategies and vaccines formulated to target these genetic mutations could improve the standard of care of COVID-19 patients. However, future studies with more uniform geographical representation across nations and correlations between the genetic variability and the clinical manifestations of COVID-19 are required to devise more efficient therapeutic policies.
Journal reference:
- Cairo A, Iorio MV, Spena S, Tagliabue E, Peyvandi F (2022) Worldwide SARS-CoV-2 haplotype distribution in an early pandemic. PLoS ONE 17(2): e0263705, DOI: https://doi.org/10.1371/ journal. pone.0263705, https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0263705
 WVU researchers explore how to restore vision in people with inherited blindness
WVU researchers explore how to restore vision in people with inherited blindness