The coronavirus disease 2019 (COVID-19) pandemic caused by SARS-CoV-2 has resulted in more than 491 million cases globally and over 6.15 million deaths.
In two years, the research community has extensively studied and characterized SARS-CoV-2 with thousands of viral genomes sequenced from COVID-19 patients.
These characterizations have been vital to understanding the evolutionary and pathogenic features of SARS-CoV-2 and designing effective therapeutics. However, there is limited knowledge on nucleotide variation and viral genome plasticity, particularly the RNA modifications caused by host cells.
In humans, adenosine to inosine (A-to-I) change catalyzed by adenosine deaminase acting on RNA 1 (ADAR1) is the most common RNA modification, and the translation machinery recognizes the resultant inosine as guanosine (G).
Moreover, the extent to which the SARS-CoV-2 genome is subject to RNA editing inside host cells remains unknown. It is crucial to evaluate whether RNA editing occurs in the SARS-CoV-2 genome because the virus uses the minus (-) strand of its RNA as the template for replication. Any nucleotide change, if induced, could cause genetic variations that subsequent generations could inherit. Besides, the genome of SARS-CoV-2 is vastly protein-coding, unlike human genomes. Therefore, any nucleotide change in the RNA would, with high probability, reflect changes in the amino acid sequences of protein.
The study and findings
The current study identified a nucleotide variant pool from meta-transcriptomic sequence reads with a bioinformatics pipeline and tested if actual RNA editing sites were present in the collection.
First, meta-transcriptomic sequence reads from the bronchoalveolar lavage fluid samples were systematically analyzed. A bioinformatics platform was developed to detect single nucleotide variants (SNVs) that could remove low-quality reads, identify viral reads with Fastv, trim end nucleotides, generate high-quality alignment, and detect SNVs with significant significance variable allele frequency (VAF).
Of the 19 samples assessed, one had no detectable SNVs; in the rest of the samples, a consistent pattern of A > G and thymidine (T) > cytidine (C) substitutions. The SNVs, A > G and T > C, correspond to the A-to-I transition in positive (+) and (-) strands, respectively, of SARS-CoV-2 RNA. The authors observed about 144 recurrent SNVs with VAF ranging from 0.5 % to 70 % and obtained a set of A-to-I candidate sequences. Analysis of sequences flanking the candidate sites revealed G depletion and enrichment preference at the editing sites’ -1 and +1 positions, respectively. RNA editing at about 55% of sites resulted in non-synonymous changes, mainly in the ORF1ab and spike protein.
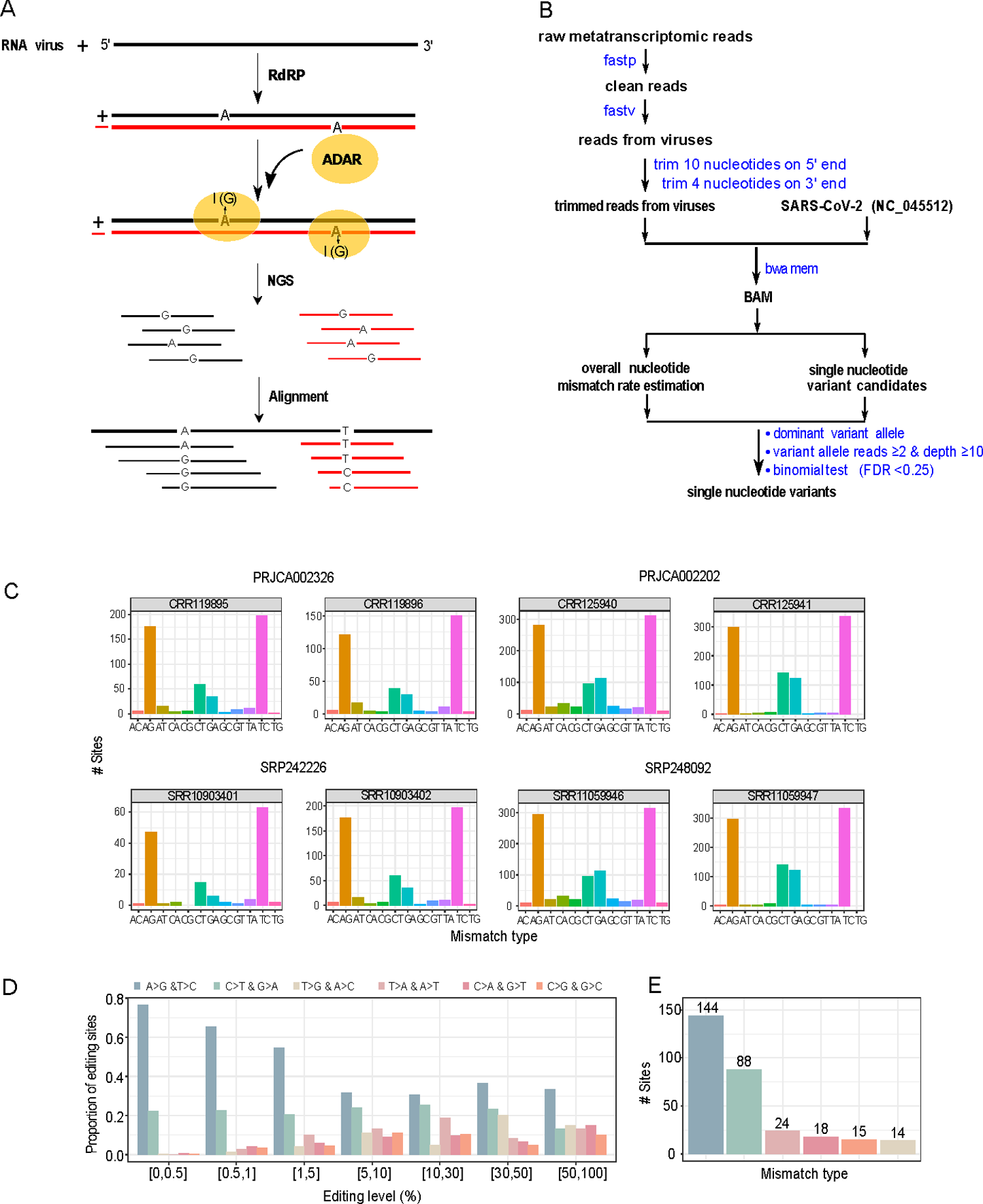
Identification of A-to-I RNA editing candidate sites from metatranscriptomic sequencing data from COVID-19 patient samples. (A) A cartoon illustration showing ADAR-mediated A-to-I RNA editing on positive and negative strands of SARS-CoV-2, which causes A>G and T>C substitutions, respectively. (B) The bioinformatics pipeline for identifying SNVs. (C) The distribution of 12 possible SNV types in eight representative samples (two per study). See the remaining samples investigated in S2 Fig. (D) The distribution of six distinct SNV types at different VAF cutoffs (paired SNV types corresponding to the same SNV changes in positive and negative strands are combined). (E) The distribution of six distinct SNV types was identified with the same procedure (VAF: 0.5%-70%, and recurrence ≥ 3 samples). https://doi.org/10.1371/journal.pgen.1010130.g001
As SARS-CoV-2 RNA replicates in the host cell cytoplasm, harboring the p150 ADAR1 isoform, the research group speculated that the inferred RNA editing sites would have a higher binding affinity to ADAR1. A hybrid neural network was built based on the ADAR1 crosslinking immunoprecipitation (CLIP) followed by high-throughput sequencing (CLIP-seq) peak set to predict binding affinity. It was noted that many RNA editing sites had a greater tendency to bind to ADAR1.
RNA sequencing (RNA-seq) data were obtained from a cell line infected with SARS-CoV-2. Ruxolitinib, an immunosuppressive agent, significantly reduced the expression levels of ADAR1 in the infected cells. The inhibition was more apparent for its p110 isoform, and RNA editing was lower at the candidate sites.
Of the 84 candidate editing sites, 75% exhibited reduced editing levels, indicating the direct effect of ADAR1 of host cells on the A-to-I viral RNA editing. RNA editing sites were enriched in clusters, i.e., the distance between RNA editing sites was shorter. CROSS, a computational RNA structure prediction algorithm, revealed that RNA editing sites had a significantly high propensity to form secondary structures. The amino acid substitutions induced by the RNA editing events were studied in the context of binding affinity of T cell epitope to human leucocyte antigen (HLA) and improved binding affinity with the edited peptide relative to wild-type peptide was noted.
Conclusions
The present study reported that ADAR1 mediates A-to-I RNA editing of SARS-CoV-2 RNA in host cells. Thus, RNA editing might represent another factor of genetic variation of the SARS-CoV-2 genome shaping its evolution and plasticity. In addition to the p150 ADAR1 isoform, the p110 isoform was also observed as mediating viral RNA editing.
The RNA editing induced by the host cell might accelerate the viral evolution. Still, its fate is dependent on the fitness effect and subject to selection pressures such as purifying selection if found deleterious and positive selection if found advantageous. One of the few limitations of the study includes the non-validation of the RNA editing sites experimentally.
 Sperm RNA aging shift that may explain paternal age effects
Sperm RNA aging shift that may explain paternal age effects