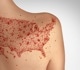
In a recent study posted to the Virological discussion forum, researchers revealed a draft genome sequence of the monkeypox virus associated with a recent outbreak in Portugal.
. Image Credit: cometa geo/Shutterstock") Study: First draft genome sequence of Monkeypox virus associated with the suspected multi-country outbreak, May 2022 (confirmed case in Portugal). Image Credit: cometa geo/Shutterstock
Study: First draft genome sequence of Monkeypox virus associated with the suspected multi-country outbreak, May 2022 (confirmed case in Portugal). Image Credit: cometa geo/Shutterstock
Background
Monkeypox is a rare disease caused by the monkeypox virus. It is a member of the same genus, including the variola virus causing smallpox, the Orthopoxvirus genus under the Poxviridae family. Monkeypox is a zoonotic disease endemic to tropical rainforest regions of West and Central Africa and is sometimes reported in other regions. The disease was first identified in a nine-year-old boy in the Democratic Republic of Congo in 1970.
The enveloped monkeypox virus is a double-stranded (ds) deoxyribonucleic acid (DNA) virus with different rodents and non-human primates as hosts. The virus has been classified into two distinct genetic clades: the West African clade and the Central African (Congo Basin) clade. Symptoms are similar to those in smallpox patients, albeit less severe. It spreads by direct contact with body fluids, lesions, contaminated surfaces/materials, or respiratory droplets.
Multiple monkeypox outbreaks have been reported in May 2022 in non-endemic countries such as the United States (US), Portugal, Spain, the United Kingdom, Belgium, and Sweden. Sequencing the viral genome would help understand its epidemiology, transmission, and sources of infection.
The study and results
In the current study, researchers determined the genomic sequence of the monkeypox virus from an infected Portuguese male. A swab sample was obtained from the patient’s lesions on the skin on May 4, 2022.
The team used a QIAamp DNA blood kit and extracted DNA from the skin exudate. A rapid barcoding sequencing kit was used to prepare libraries, and subsequently, shotgun metagenomics sequencing was carried out using Oxford Nanopore MinION apparatus. They mapped reads to a reference sequence using the ‘inside the FLU’ (INSaFLU) web suite and manually inspected the sequence to validate the position of variants. Phylogenetic analysis was conducted using the new draft sequence with publicly available genomic sequences.
Phylogenetic analysis revealed that the sequence belonged to the West African clade. They observed that the virus was most closely related to viruses associated with previous exportations from Nigeria to different countries in 2018 and 2019. The draft sequence covers approximately 92% length of the reference sequence. The authors reported that the draft sequence would soon be curated. The regions with lower coverage, insertions/deletions (indels), or homopolymeric tracts would be refined when ongoing sequencing data from the Illumina platform becomes available.
 Why measles remains one of the most contagious viral diseases worldwide
Why measles remains one of the most contagious viral diseases worldwide