 *Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Background
The identification and nomenclature of pathogenic lineages are important from the perspectives of treatment, disease-related communications, and research. The nomenclature systems for pathogens are generally based on genotype, phenotype, and geography.
Serology or antibiotic vulnerability are the two phenotypes used to classify pathogens. Comparatively, a nomenclature based on geography applies to pathogens that have non-human species as reservoirs, such as the Zaire Ebola and Chikungunya viruses.
Genotype-based classification focuses on the resolution of pathogens in a phylogenetic tree into reciprocally monophyletic or exclusive clades, with the samples in a clade inferred to have descended from a common ancestor. This method had been used for the respiratory syncytial virus, influenza virus, and dengue virus.
The Phylogenetic Assignment of Named Global Outbreak (Pango) nomenclature system, which is a genotype-based system of classification, uses crowdsourced lineage proposals and has provided the names for all the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern.
However, since Pango requires the lineages to be manually curated and designated, the increasing volume of data and diminishing public investments, as well as the personal and regional bias in assessing the importance of different mutations, highlight the need for a more streamlined and objective metric to designate emerging pathogenic lineages.
About the study
In the present study, researchers propose a heuristic approach to define and expand genotype-based nomenclature, which uses branch lengths from phylogenies scaled to genetic distance. This method can expand existing pathogen nomenclature systems and generate new nomenclature for emerging pathogens.
The proposed method is optimized to use the haplotype information at the sample level and can be applied to large phylogenetic trees to produce a hierarchical arrangement of distinct lineages. A series of user parameters can also be incorporated to allow researchers and epidemiologists to weigh critical lineage definition elements and tracking efforts for each pathogen.
The utility of this method was evaluated using the global SARS-CoV-2 phylogeny. Based on its success, the researchers collaborated with Pango researchers to incorporate this method into the existing SARS-CoV-2 lineage nomenclature system.
Furthermore, the potential for the use of this method for other pathogens was evaluated by testing it against the Chikungunya virus and Venezuelan equine encephalitis (VEE) virus complex phylogenies.
Results
The new heuristic approach-based method was efficient in analyzing extremely large phylogenies. When tested using the latest global SARS-CoV-2 phylogeny, this approach produced results that were similar to existing lineage designations based on the Pango classification system.
The genotype representation index (GRI), which is calculated based on the number of descendants from a node, was defined as the branch length between the node and parent lineage or root of the phylogenetic tree, as well as the branch length between the node and each tip or descendant. Therefore, as long as the branch lengths are based on genetic distance, GRI can be calculated for any rooted phylogeny.
Lineage designation is based on high GRI values at internal nodes. The proposed method uses an iterative process to define hierarchical lineages. Furthermore, epidemiologically important elements, such as international transmission and protein sequence changes, can be used for a weighted calculation of the GRI.
The application of this nomenclature system to Chikungunya and VEE phylogenies revealed that while the automated lineage designation corroborated the existing Chikungunya nomenclature system that was based on geography, the VEE nomenclature system, which was based on serology, was paraphyletic in the automated nomenclature system and did not form any clades.
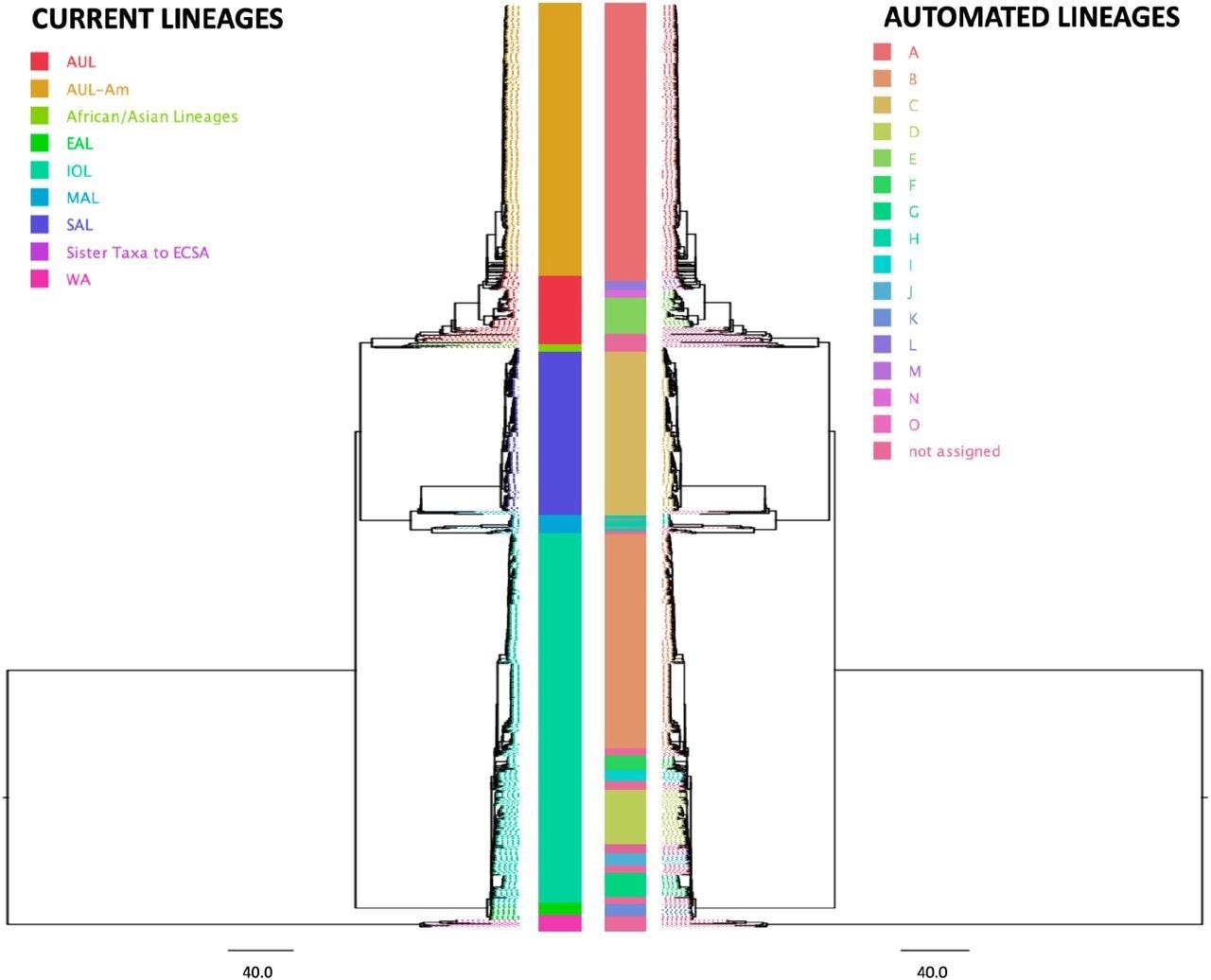 Figure 3: Comparison of the geography lineage designation (left tree) with automated lineage designation (right tree) of Chikungunya virus, based on a tree previously generated by the Augur pipeline (Huddleston et al 2021) and visualized on FigTree v.1.4.4.
Figure 3: Comparison of the geography lineage designation (left tree) with automated lineage designation (right tree) of Chikungunya virus, based on a tree previously generated by the Augur pipeline (Huddleston et al 2021) and visualized on FigTree v.1.4.4.
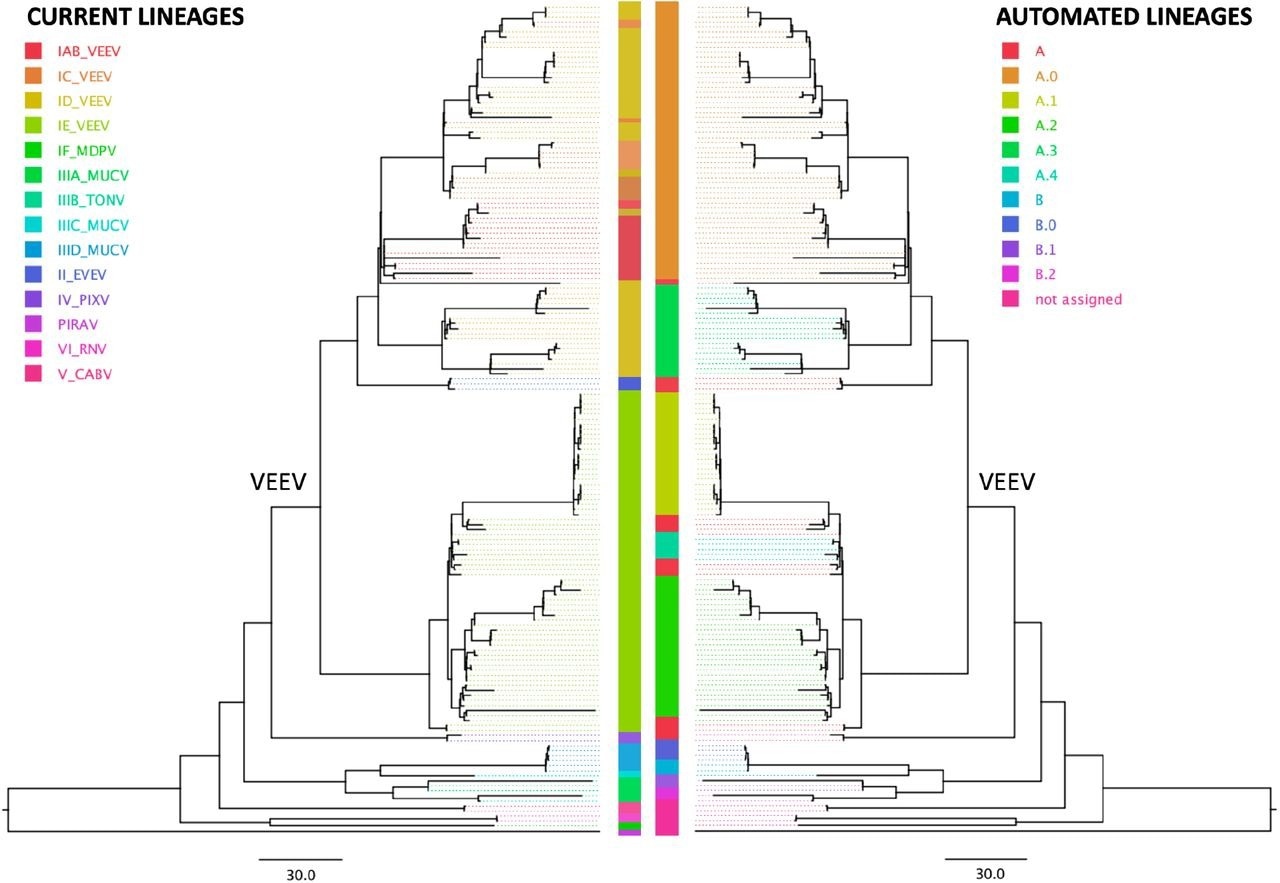
Comparison of the serology subtype designation (left tree) with automated lineage designation (right tree) of the Venezuelan equine encephalitis virus complex (VEE), based on a tree previously generated by the Augur pipeline (Huddleston et al 2021) and visualized on FigTree v. 1.4.4. According to the current nomenclature, VEE encompasses Everglades virus (EVEV), Mucambo virus (MUCV), Tonate virus (TONV), Pixuna virus (PIXV), Cabassou virus (CABV), Rio Negro virus (RNV), Mosso das Pedras virus (MDPV), Pirahy virus (PIRAV) and the Venezuelan equine encephalitis virus (VEEV). The VEEV clade is labeled in the tree.
Method limitations
Since the classification system is based on phylogenies, new data and optimization of phylogenetic trees can change the relationship between lineages and invalidate existing lineages. However, given that there is currently no system or universal definition for classifying taxa below the species level, this method provides a uniformly applicable and streamlined method for classifying pathogens.
Conclusions
Overall, the results suggest that the automated lineage designation system presented in this study provides a flexible, generic, and streamlined method to expand existing pathogen nomenclature systems and define lineages for emerging pathogens using phylogenies and branch length information.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Journal reference:
- Preliminary scientific report.
McBroome, J., de Bernardi Schneider, A., Roemer, C., et al. (2023). Automated Agnostic Designation of Pathogen Lineages. bioRxiv. doi:10.1101/2023.02.03.527052
 COVID-19 immunity stays specific and barely boosts protection against common cold coronaviruses
COVID-19 immunity stays specific and barely boosts protection against common cold coronaviruses