Human health status can be determined based on the circulating metabolite levels. Although it is important to understand the genetic architecture of these metabolites, to date, they are not well understood. Considering this research gap, a recent Nature Communications study focused on performing whole-genome sequencing analysis of circulating human metabolites in a multi-ethnic population.
Circulating metabolites are heritable and play a vital role in health outcomes. Several genetic disorders dysregulate blood and tissue metabolite levels, leading to various complex diseases. Recently, scientists have focussed on profiling human metabolites to assess disease outcomes.
The majority of genetic studies based on human metabolome are based on European populations, however, only a few have considered African-American and Hispanic populations. It is important to include an ethnically diverse population as it can lead to greater genetic discovery linked to varied diseases. Even though most studies have concentrated on autosomal chromosomes, it is vital to explore X chromosomes as well as better understand the genetic architecture of metabolites.
About the Study
The current study performed whole-genome sequencing (WGS) to detect genetic loci associated with 1,666 circulating metabolites in a multi-ethnic population. A total of 15,660,619 common, low-frequency, and rare variants belonging to autosomal chromosomes and the X chromosomes linked to the circulating metabolites in up to 11,840 participants were analyzed.
The participants belonged to African-American (AA), European American (EA), and Hispanic (HIS) ethnicity, who participated in the Atherosclerosis Risk in Communities (ARIC), Framingham Heart Study (FHS), Hispanic Community Health Study/Study of Latinos (HCHS/SOL), Multi-Ethnic Study of Atherosclerosis (MESA) and Cardiovascular Health Study (CHS) studies. All participants were around 57 years of age, and 57% of the cohort were women,
For replication analysis, independent participants from Jackson Heart Study (JHS), FHS, TwinsUK, and Women’s Health Initiative (WHI) studies were obtained, which comprised AA and individuals of European ancestry. A total of 18,085 individuals were used for replication analysis.
Study Findings
Using WGS, a total of 75 novel replicated metabolite-genetic locus associations were detected. Among these, 22 associations were driven by nonsynonymous variants. Gene-centric rare variant analysis was conducted for a subset of metabolites, identifying 126 gene-metabolite pairs. This finding indicated the associations between 45 metabolites and 105 genes.
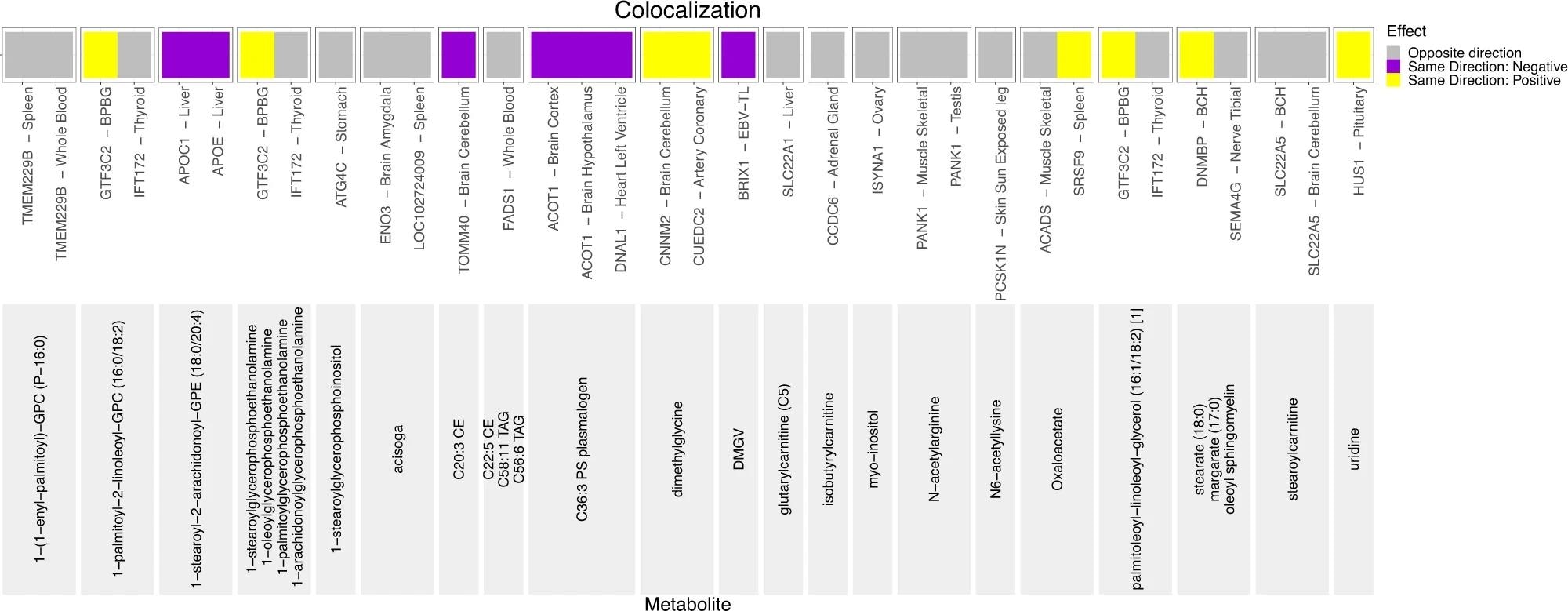 The direction of the effect of minor allele on metabolite levels and gene expression is shown in the legend. At the bottom of the graph, in light gray, are the names of the metabolites. Above the names of metabolites are eQTL gene-tissue pairs. If both the effect of minor allele on metabolite levels and on gene expression is more than 0, such variant-metabolite-gene eQTL combinations are marked in yellow and annotated as “Same Direction: Positive”. If the effect of minor allele on metabolite levels and on gene expression is less than zero, such variant-metabolite-gene eQTL combinations are marked in purple and annotated as “Same Direction: Negative.” If the effect of minor allele on metabolite levels is less than zero and the effect of minor allele on gene expression is more than zero, or vice versa, such variant-metabolite-eQTL combinations are marked in gray and annotated as “Opposite Direction”. Additionally, the following acronyms were used for tissues: BPBG brain putamen basal ganglia, BCH brain cerebellar hemisphere, EBV-TL - Cells EBV-transformed lymphocytes.
The direction of the effect of minor allele on metabolite levels and gene expression is shown in the legend. At the bottom of the graph, in light gray, are the names of the metabolites. Above the names of metabolites are eQTL gene-tissue pairs. If both the effect of minor allele on metabolite levels and on gene expression is more than 0, such variant-metabolite-gene eQTL combinations are marked in yellow and annotated as “Same Direction: Positive”. If the effect of minor allele on metabolite levels and on gene expression is less than zero, such variant-metabolite-gene eQTL combinations are marked in purple and annotated as “Same Direction: Negative.” If the effect of minor allele on metabolite levels is less than zero and the effect of minor allele on gene expression is more than zero, or vice versa, such variant-metabolite-eQTL combinations are marked in gray and annotated as “Opposite Direction”. Additionally, the following acronyms were used for tissues: BPBG brain putamen basal ganglia, BCH brain cerebellar hemisphere, EBV-TL - Cells EBV-transformed lymphocytes.
Mendelian Randomization exhibited a total of 13 metabolites associated with the risk of 12 phenotype outcomes, such as macular degeneration and type 2 diabetes. In addition, 16 metabolites were linked to 29 protein quantitative trait loci (pQTLs).
In contrast to previous research, this study demonstrated a contemporary approach to investigating cross-platform harmonized metabolite levels in pooled samples. This approach improved computational efficiency. The new approach was able to reproduce hundreds of known metabolite loci, control genomic inflation, and enabled the identification of novel genes through rare variant analyses. Further, the benefits of joint analyses, particularly for large genomic datasets, were highlighted in this study.
The authors claim this piece of research to be the first to establish a metabolite genetic association using multi-ethnic populations. Here, additional insights associated with the biological pathways were provided, which were uncovered by investigating the interacting effects between metabolites and proteins.
Even though pooled sample analysis is computationally efficient, the variant-set test could be intensive and costly for whole genome analysis. In the current study, gene-centric rare variant analyses among 230 metabolites were initially performed, which indicated significant common variants. These metabolites exhibited relatively high heritability, which was exploited to study the impact of rare variants on these metabolites.
Versatile analytic approaches, such as colocalization and pathway analyses, were used to determine possible mechanisms associated with the replicated novel findings. A total of 18 unique loci were identified using colocalization analyses. This analysis led to the detection of the novel replicated variant colocalized with the eQTL for 26 unique genes in the Genotype-Tissue Expression (GTEx) tissues, indicating the biologically plausible genes. For instance, the splice site intronic SLC22A1 variant was found to be associated with increased levels of the lysine metabolism metabolite glutarylcarnitine (C5). Similarly, replicated intronic ELL variant rs8109573 was found to colocalize with decreased expression of ISYNA1 in ovaries.
In the context of biological pathways, the function of acylcarnitines in relation to coagulation was observed. This is noted to be the first genetic evidence linking metabolites to blood coagulation. The MR analyses helped detect the bi-directional associations between proteins and metabolites.
Conclusions
The current study demonstrated the possibility of performing computationally efficient pooled analysis using WGS and metabolomics data. This approach could be used in future projects as well. Notably, the genetic architecture of circulating metabolites in a multi-ethnic population was determined based on comprehensive functional annotation and common and rare variants. In addition, causal relationships between the genes, various phenotypes, metabolites, and plasma protein levels were determined.
 Research reveals potential new strategy for targeting deadly small cell neuroendocrine cancers
Research reveals potential new strategy for targeting deadly small cell neuroendocrine cancers