They review over 80 publications on critical mechanisms, including synaptic dysfunctions, oligomers of amyloid beta (Aβ) protein, protein clustering and membrane structure alterations, and α-synuclein aggregation. Their findings suggest that altered cholesterol synthesis and metabolism are shared features of most investigated neurodegenerative diseases. While cholesterol-lowering drugs can partially reduce these diseases' risk, additional research is required to develop future targeted pharmacological interventions against these conditions.
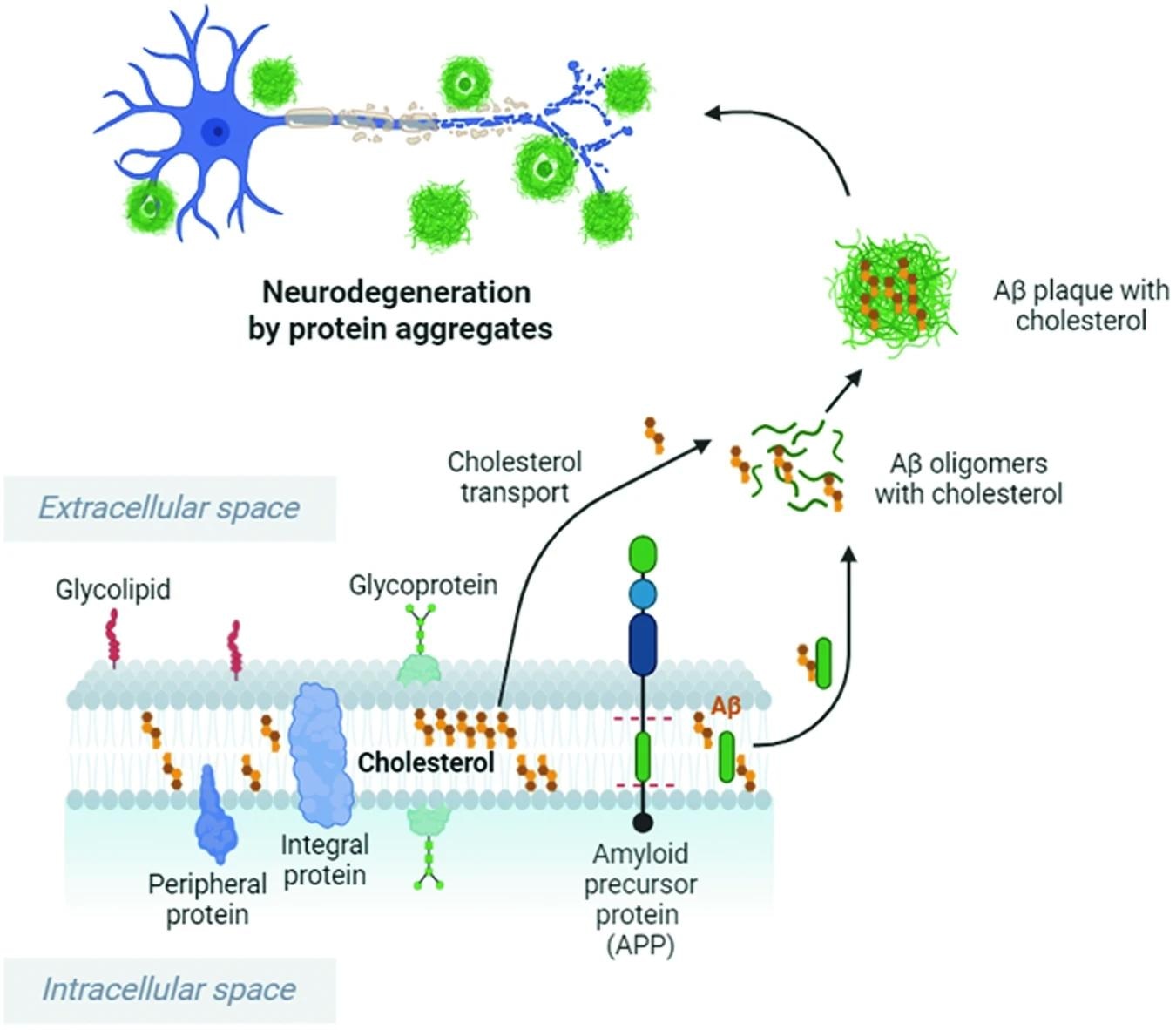
Cholesterol enhances and accelerates APP cleavage by Bace1, leading to increased Aβ oligomer and plaque formation. Cholesterol binds to Aβ and increases the resistance of Aβ fibrils and oligomers to degradation. Cholesterol imbalance and high extracellular cholesterol levels can stimulate the production and accumulation of Aβ peptides, which cause Aβ oligomer formation and aggregation in the brain, resulting in neuronal damage. This image was created with BioRender.com. Study: Cholesterol imbalance and neurotransmission defects in neurodegeneration.
Background
Cholesterol is a waxy, fat-like substance found in the cell membranes of all human cells. It plays an integral role in neuronal signaling and synaptic connections, especially in the brain. Notably, the brain contains between 20-25% of all the body's cholesterol reserves, making it the organ with the highest cholesterol concentration in the human body. Interestingly, peripheral cholesterol (cholesterol absorbed from the diet that circulates in the bloodstream) is incapable of crossing the blood-brain barrier (BBB). Consequently, almost all of the brain's cholesterol reserves arise from de-novo synthesis (in the glia and neurons).
As humans age, the efficiency of their cholesterol-synthesizing glia and neurons diminishes, reducing their brains' cholesterol reserves and resulting in impaired synaptic plasticity and overall synaptic loss. These losses have been suggested to contribute strongly to increased risks of neurodegenerative diseases, including Alzheimer's disease (AD), Huntington's disease (HD), and Parkinson's disease (PD). Unfortunately, the molecular and pathological mechanisms underpinning these observations remain understudied.
About the study
The present study collates and reviews more than 80 publications on cholesterol to elucidate four key molecular mechanisms underpinning the associations between cholesterol imbalances and subsequent adverse neurodegenerative outcomes. These mechanisms include: 1. Synaptic dysfunction, 2. Amyloid beta (Aβ) aggregation, 3. Protein clustering, membrane structure alterations, and 4. α-synuclein (α-syn) aggregation.
Molecular mechanisms
1. Synaptic dysfunction – Cholesterol has been observed to comprise up to 80% of the plasma membrane of synapses and is essential in both their formation and function. Research has highlighted that cholesterol imbalances can significantly alter the ability of synapses to share neurotransmissions effectively, eventually resulting in neurodegenerative diseases. Molecular models have revealed that cholesterol imbalances adversely impact Ca2+-dependent vesicle fusion, altering membrane elasticity. In extreme cases, this can result in significant unwanted membrane bending and curvature alterations, increasing the energy required for membrane/vesicle fusion and impairing neurotransmission.
2. Oligomers of Aβ protein – The amyloid precursor protein (APP) is converted into Aβ protein via enzymatic cleavage catalyzed by β-secretase Bace1. Given its integral role in protein aggregation and folding, the successful conversion of APP to Aβ protein relies on normal cholesterol levels in the brain, with alterations in the latter observed to cause misfolding in the former. Misfolding of Aβ protein results in forming Aβ plaques, the accumulation of which is a hallmark of AD pathology. "Cholesterol imbalance and elevated extracellular levels of cholesterol can promote the production and accumulation of Aβ peptides, which induce the formation of Aβ oligomers in the brain, thus contributing to neuronal damage and cognitive decline." Tau (specifically, hyperphosphorylated tau) aggregation, another hallmark of AD pathology, is also contingent on cholesterol concentrations, given the membrane curvature properties of the latter. Recent research has elucidated that cholesterol-free membranes cannot form tau fibrils, while membranes containing cholesterol influence tau fibril formation contingent on cholesterol concentration and its associated membrane curvature. Unfortunately, the impacts of cholesterol on tau nucleation remain understudied and currently unknown.
3. Protein clustering and membrane structure – Cholesterol has been shown to play integral roles in regulating normal membrane curvature, structure, and fluidity. Curvature and deformation are essential for vesicle function and fusion pore stabilization, enabling neurotransmitter propagation across the central nervous system. Recent research has further revealed that cholesterol is critical in the protein clustering and intracellular organization of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins. Communication between different SNARE proteins (syntaxin-1A, SNAP-25, and VAMP-2) together comprises the core SNARE complex, which mediates vesicle fusion and, in turn, neurotransmitter release within a synapse.
4. α-syn aggregation – The genesis and progression of PD are characterized by the accumulation of misfolded α-syn proteins in Lewy bodies (LBs). The mechanistic underpinnings of this process are a consequence of α-syn binding to membrane lipids. Imbalanced cholesterol accelerates α-syn aggregation and LB formation, increasing PD risk.
Future therapeutic interventions
The most essential neural protein family involved in the transport and normal metabolism of cholesterol is the apolipoprotein E (ApoE) family (ApoE2, ApoE3, and ApoE4). ApoE4 has further been identified as a crucial risk factor in last-onset AD. Unfortunately, the molecular contributions of the ApoE family are poorly understood and require further investigation. However, given the role and importance of ApoE4 in both cholesterol homeostasis and AD pathology, it may be highlighted as a target for clinical trials and future pharmacological interventions.
Conclusions
The mechanistic underpinnings of cholesterol on various neurodegenerative diseases are context-dependent. However, the present study highlights how imbalances in cholesterol levels, especially in the brain, can increase the risk of these diseases and suggests potential strategies for their management.
"While cholesterol-lowering drugs, e.g., statins, have shown some potential in reducing the risk of certain neurodegenerative diseases, further research is required to fully understand the role of cholesterol and develop targeted therapeutic interventions."
 Discovery of fat-regulating enzyme points to a new obesity therapy
Discovery of fat-regulating enzyme points to a new obesity therapy