Table of Contents
Abstract
Introduction
Definitions
Sources
Regulations And Guidance
Clinical Trial Material: The Next Milestone in E&L Research
Requirements for Clinical Trial Material
Which Products Get Queries?
Category 1: Products With a Material Qualification Program
Category 2: Vendor Supplies Package of Extractables for a Standard Composition
Category 3: Lack of Extractables Data for a Non-Standard Product
Category 4: Parenteral Dosing Devices (Pumps, Infusion Sets, IV Bags)
Challenges of Planning an Early-Phase Program
Condensed Early-Phase E&L Study Design
Case Study 1: Non-Aqueous Vial/Stopper
Case Study 2: Infusion Set
Conclusions
Abstract
Not long ago, the assessment of extractables and leachables in drug products was performed mainly during late-phase research. Recently, regulators have been increasingly asking for additional E&L information for early phase clinical data, especially if the products are lacking an extractables package provided by vendor or if they are non-standard. The health authority requests can often be time-consuming and expensive, and could potentially delay the clinical trials.
It has become possible to identify the products and attributes which are more likely to trigger queries from the FDA through early phase research. A streamlined E&L design which helps the sponsors answer or potentially avoid queries for early phase research have been described. The study design helps sponsors to proactively gather this information. It also helps build Quality by Design (QbD) approach in the drug development process and reduces the resources needed for development.
Introduction
Leachable compounds from manufacturing equipment, container closures, and delivery systems can adversely affect drug potency and patient safety. E&L evaluations have progressed and assumed a greater role in drug development process over the last 20 years. Initially, E&L studies were needed for orally inhaled and/or nasal drug products (OINDPs), later for parenterals, dermal and ophthalmic products. Since the beginning of single use systems (SUS), E&L studies drastically have increased for manufacturing equipment because of the presence of plastic components which replace traditional stainless steel components. Each of the evolutions has focused on final approval and registration of a drug (e.g. NDA, ANDA, BLA), and not on the any information on what may be needed during early development phase of the process.
A general guideline has been provided by FDA about extractables and leachables, however what should be measured has not been clarified. Guidance is also lacking on how to measure extractable and leachable compounds and the safe levels of such compounds. Therefore, industry groups are relied upon as the main source for advice on the best practices for successful registration of the products.
In last five years, there has been a significant rise in the requests from FDA for complete E&L data of Phase 1 and Phase 2. In addition to the clinical trial data, the requests also cover dosing components and manufacturing equipments. Until such data were made available by the sponsors, many programs were kept on hold, which lead to high costs and overall delay.
Considering the time and expenses incurred in conducting E&L studies, it can be an arduous burden on drug development. Professionals from analytical development, CMC, and manufacturing and processing units often struggle with the cost and time component of E&L information acquiring process.
Fortunately, with observation it has become possible to identify few commonalities in queries regulators raise and the types of programs which receive such requests for E&L data. It mainly involves unique syringe configurations, non-aqueous formulations, drug delivery systems and biomedical devices. E&L studies are more likely to be needed at an early phase of development if there are proprietary packaging, uncommon components, when the vendor has not supplied E&L data, and when components are not yet approved.
In response to the rising need for brief and cost-effective methods for E&L studies, we created a compressed experimental design for to recognize extractables and leachables in early phase development. We have employed it successfully for a variety of drug products, which has helped drug development teams in answering or avoiding regulatory queries.
Definitions
Extractables: Compounds which transfer out of materials under harsh laboratory conditions.
Leachables: Compounds which transfer into the product under actual usage situations, such as production step. Normally a subtype of extractables.
Extractable ≠ Leachable: Extractables don’t necessarily leach always, leachables don’t necessarily extract always.
Safety Concern Threshold (SCT): The quantity below which there exists a negligible toxicity risk depending on the dosing. It is mainly utilized in case of leachables with unknown safety profile; compounds with known safety profile have toxicological data for reference.
Analytical Evaluation Threshold (AET): The quantity above which a leachable or extractable requires to be reported for potential toxicity evaluation. It depends on the SCT.
Sources
- Manufacturing equipment
- Syringes
- Vials
- Foil overwraps
- Primary packaging componentsMedicine droppers
- Inks and dyesSecondary packaging components
- StoppersLabels
- Shipping materials
- Infusion sets
- IV setsAssociated/dosing components
SUSProcessing components
Regulations And Guidance
Similar requirements are laid out by FDA has for equipment and container closure systems, for both biologics and small molecule drugs. Till now, other than the need to carry out E&L studies, FDA has provided only minimal additional guidelines:
- Guidance for Container Closure Systems for Packaging Human Drugs and Biologics (1999)
- Reviewers Guidance for Nebulizers, Metered Dose Inhalers, Spacers and Actuators (1993)
- Metered Dose Inhalers (MDI) and Dry Powder Inhalers (DPI) (1998)
- Nasal Spray and Inhalers Solution, Suspension, and Spray Drug Products (2002)
- Drug Products Packaged in Semipermeable Container Closure Systems (2002)
Due to the lack of guidance by regulatory authorities, regarding the objectives, endpoints and conduct of the studies, sponsors are largely dependent on best practices and industry groups for advice. These include USP, ICH, PQRI (best-practices for parenterals, inhalation devices, and ophthalmics), BPSA, and BPOG (guidance on SUS and bioprocessing equipments).
Collectively, these guidelines deal with the information needed to submit a registration application for PLA, NDA, or NADA, but lack the guidance on requirements for early development phase.
Clinical Trial Material: The Next Milestone in E&L Research
A sponsor usually starts E&L studies at the end of Phase 2 trial, as the container closures are chosen by then, and the production process is being confirmed. This is followed by generation of stability data for leachable during Phase 3. A complete E&L program can take time ranging from six to eighteen months to create the data needed for registration. This makes it tough to do a complete E&L analysis during a shorter length of Phase 1 and Phase 2 studies.
We noticed the first query regarding E&L data for Phase 1 in 2012, for a non-aqueous formulation utilizing a non-standard vial/stopper system. Since then, we have studies a huge number of programs which received regulatory requests for E&L assessments and/or data of Phase 1 or, in few cases pre-IND. In some of the cases, FDA mentioned that USP testing was inadequate for carrying out a risk assessment. A significant sum of these programs was kept on clinical hold awaiting data generation.
Requirements for Clinical Trial Material
While specific requirements by health authorities are not very clear, Dr. Ingrid Markovic from CBER said in a presentation in 2014, “E&L and other impurities are deemed to be controlled in the IND phase because the clinical outcomes are closely monitored; therefore, E&L studies are generally not required, unless so deemed warranted. However, from a manufacturing perspective, it is advisable to be cognizant of E&L during component and packaging selection in early development to avoid possible problems in late development.”
These comments highlight the challenges faced while performing E&L analyses during early phase. Typically, while it might be advisable to have the data for future improvement in quality, it is not mandatory for safety evaluation “unless so deemed warranted.” The important part is, what do health authorities imply by “deemed warranted?”
Which Products Get Queries?
Based on internal data for several drug products at various drug development stages, and supplemented by information from pharmaceutical companies and colleagues, we can predict whether a given product is likely to get an E&L data query from FDA during Phase 1.
Products receiving queries tend to be in the high-risk class identified by FDA guidance document for container closure systems. Inhalation products are exception to this general pattern, probably because the manufacturers provide information to FDA beforehand.
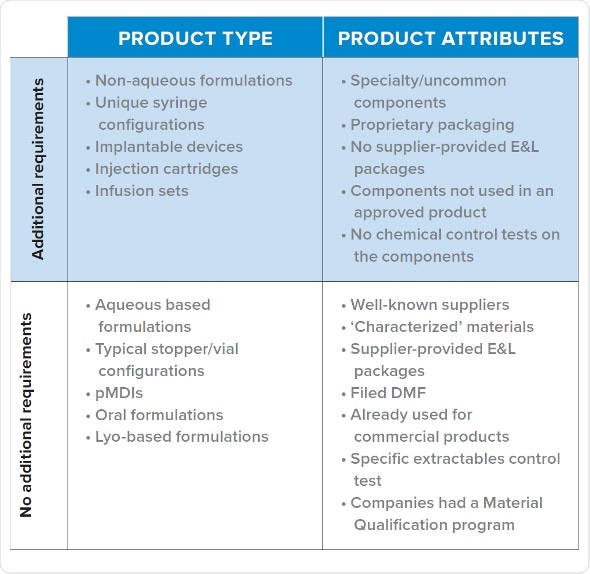
Category 1: Products With a Material Qualification Program
Products which have been evaluated for safety and quality using a material qualification program usually receive no queries for additional E&L analysis. A sponsor’s material qualification program institutes a standardized testing procedure to evaluate all the risks. The extraction employs up to ten solvents, a pH range and ionic strength, changes the ratio of organic/aqueous solvents and utilizes common excipients such as surfactants in order to simulate the process conditions. Further to that, they are assessed with a variety of methods, such as HPLC. It permits comparison of possible parts (e.g. stoppers) for leachables and for their compatibility.
The standard analysis comes with a minimal risk of an unknown leachable showing up later on. It needs substantial amount of time and cost to be spent beforehand to establish the resources and create data for maintaining and performing analysis. It is efficient for the companies with strong pipelines, and those which might have many vial/stopper configurations to analyze.
Category 2: Vendor Supplies Package of Extractables for a Standard Composition
If a product did not pass a material qualification program, but the vendor provides a complete extractables profile similar to commercialized products, the sponsor is unlikely to receive a query regarding E&L data. The information which needs to be collected before selecting material consist of checking the presence of a DMF, checking prior use of the component in a successful filing, and type of analysis performed on components for approval.
This is the suggested direction for early phase drugs. Vendors should be analyzed to judge which one provides the best package. If two components are similar in their functions, it is reasonable to choose the one with better extractables package because of lower risk involved. It is also recommended to pick well-characterized materials. The one limitation is that there can be unknowns beyond the SCT level.
Typical design which manufacturers employ to create data needs a lot of effort. As with Category 1, leachable data is not usually needed as extractables are considered worst-case leachables. If results of leachable studies are fruitful, the extractable methods of vendor might be available. Some CROs which do E&L analyses have in-house generic methods for screening which can be utilized. It should be considered that extractable unknowns could appear as leachables, although data from vendor should identify such ‘bad actors.’
Category 3: Lack of Extractables Data for a Non-Standard Product
Non-standard products usually receive queries from CDER and CBER and for early phase data. Even though, each product which receives a query for additional E&L data falls into this category, not every product which falls in this category needs additional E&L testing. The sponsor does not have a program for material qualification, it was not given with an extractables package by vendor, and the drug is a non-standard formulation. Proactive creation of early phase data may benefit a sponsor expecting queries from FDA.
Category 4: Parenteral Dosing Devices (Pumps, Infusion Sets, IV Bags)
An exception to the above scenarios are infusion sets employed in clinical studies. They tend to trigger queries from FDA to provide additional E&L data. Products which are dosed via infusion during clinical studies are also at very high risk of being put on hold if E&L queries are not addressed.
Challenges of Planning an Early-Phase Program
Designs for early phase research should be shortened to account for the cost, time, and resource limitations typically associated with early phase studies. A complete E&L testing may take more than eighteen months and cost up to $100,000-500,000, which is not in-line with the expected duration and budget of typical early phase program. So how does one design a study which is scientific and focused on safety, while also addressing the cost and time limitations of early phase research?
Condensed Early-Phase E&L Study Design
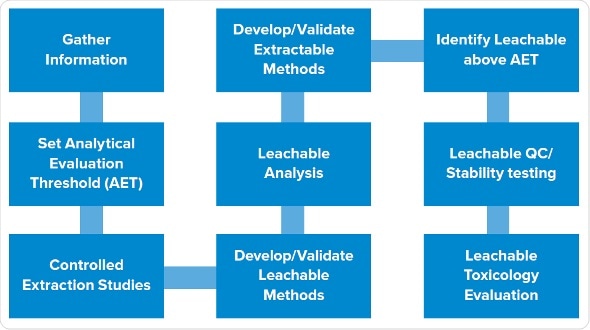
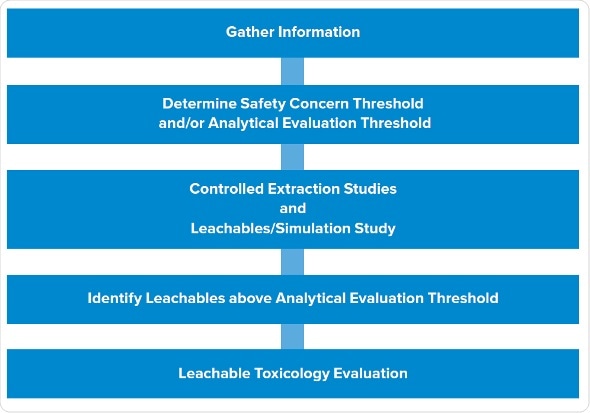
The typical, late-phase E&L design starts with gathering information, followed by determination of SCT/AET, studies of extractables, and studies of leachables. We have trimmed this design to render it phase appropriate, while still giving high-quality data which lowers risk and solves the potential queries which a company may get from the agency. We have utilized this design successfully numerous times, especially for the products put on hold.
The first two steps remain same: gather information and establish analytical level. The third step involves leachable and extraction studies in parallel instead of in stepwise manner.
Process for conducting an Extractables and Leachables study
Process for conducting an early-phase Extractables and Leachables study.
It allows to assess all extraction samples, and dummy or actual leachable samples to create that data in parallel. Then we recognize potential leachables in all the samples which are above AET. The final step is to carry out toxicity evaluations of the leachables.
Extractables program: We carry out asymptotic extractions according to the typical program with at least three solvents, covering the pH range and polarity of the product. One of the solvents mimics the placebo. We use sonication or reflux, ensuring not to be too harsh as it can create spurious extractables not likely to be leachables. Extractables are evaluated using gas chromatography, HPLC , ICP for metals etc..
Leachables program: Early phase products usually have shorter expiry dates, often around one month, and infusion sets may be used one or two days only during a study. This allows the reduction the experimental design for simulating a faster leachables profile.
Drug product and placebo are stored in the container closure system to be assessed under accelerated and normal conditions. Then we age or stress the samples in parallel, or start the leachables even prior to starting the extraction studies, to create leachable and extractable samples which we can assess concurrently. In addition to that, we assess placebos and forced degradation of active substances.
As everything is carried out in parallel, subtractive fingerprinting of different samples is carried out for each technique. For example, HPLC profiles are created for leachables and extractables in the active substances. Forty peaks of extractable may be observed, but only three among them show up in the leachables. This means that we do not have to recognize all 40 extractables as the data shows that 37 of them do not leach. Instead, we only focus on the three which showed up in the samples.
Analytical interpretation: Data interpretation is simple, comparing fingerprints between placebo and extraction samples. Use of active substances and formulation samples permits removal of peaks associated with active substances and drug products. In this manner, we can trace back peaks in drug product to identify whether they are of the active substances, the drug formulation or of the leachables. Then we can only focus on the peaks which can be traced back to the extractables, validating that they are the actual leachables.
Case Study 1: Non-Aqueous Vial/Stopper
This compact design was used to assess a vial/stopper system in which stopper was non-standard, had no vendor-provided information on extractables and utilized an uncommon cottonseed-oil formulation. FDA queried for E&L data before the clinical trial, which was supposed to be two months in length.
Study design: We used extractables on stopper. Leachables were created using only placebo because of two reasons: active substance was not available in adequate quantities; the active substance concentration in the product was low enough for us to assume, that it would not alter the leaching profile. Samples were stored for two months at storage temperature of 5 °C and at room temperature (25 °C). We then evaluated the leachable samples simultaneously with extractables, active substance, and drug product.
Results: There was a huge number of extractables expected since one of the solvents was cottonseed oil. Many were unknowns. Fingerprinting showed only two leachables beyond SCT and both corresponded to the known extractables. Four leachables were seen at 25 °C, out of which one was an unknown which we recognized using a library match with a gas chromatography mass spectrum. Since these all were knowns with available toxicity data, risk assessment depicted no safety issues.
It took three months from start of the program until the issuance of the report. The regulator’s request was satisfied and no additional queries were raised.
Case Study 2: Infusion Set
Assessments of drugs infused using pump in clinical trials usually focus only on drug product compatibility, evaluating whether the active substance sticks to polymer or not, and whether it loses potency or not. For this early phase study, drug was infused using a syringe pump for not more than 24 hours, but, no E&L results were submitted in the IND. The FDA raised queries about the infusion sets, but not the syringe pump or syringe. They kept the study on hold till an E&L testing was carried out.
Study design: Extractions were carried out on individual parts of the infusion set using the placebo and drug product. We dynamically created leachables under actual conditions while using the infusion pump for 24 and 48 hours at 37 °C which resembles body temperature.
Results: Provided that infusion sets were made up of five types of polymers, multiple extractables were identified, of which many were not known. Fingerprinting revealed that, even after 48 hours, no leachables were seen above AET, as anticipated because of the low contact duration. Risk evaluation showed no safety concerns even when used for twice as long as the typical use. It took six weeks for the study completion and issuance of the report, the data was acknowledged and the hold was revoked.
Conclusions
Regulatory inspection of leachables and extractables has been growing for clinical study materials. Leachables and extractables evaluations are now integral part of all stages of drug development. Luckily, we have been able to recognize the specific early phase clinical trial products and their particular aspects which render them more likely to face increased scrutiny from FDA for E&L queries.
To meet these increasing requirements for phase-suitable E&L assessments, an abbreviated design has been established which is ideal for creating data in an economic, time-efficient way for early phase clinical trial products. This experimental procedure has an added advantage of building QbD doctrines in the drug development during early phases, thereby lowering overall risk and guaranteeing patient safety. The study design aids sponsors reply or, in few cases, altogether avoid queries from FDA during Phase 1 and Phase 2 which could lead to a hold being put on a study.
Two case studies described above symbolize the many sponsors for which we have effectively utilized this compact plan to help their drug development process.

This information has been sourced, reviewed and adapted from materials provided by EAG Laboratories.
For more information on this source, please visit EAG Laboratories.