Analyzing mutations in the genome of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) enables scientists to understand the ongoing evolution of the virus.
These analyses can provide information on the degree and dynamics of these mutations, as well as assist in identifying which sites within the viral genome are vulnerable to specific mutations like purifying selection. Taken together, this information is essential for the establishment of adequate and proportionate public health guidelines on isolation, quarantine, and vaccination.
 Study: Ongoing global and regional adaptive evolution of SARS-CoV-2. Image Credit: successfulalexey78 / Shutterstock.com
Study: Ongoing global and regional adaptive evolution of SARS-CoV-2. Image Credit: successfulalexey78 / Shutterstock.com
In an effort to understand the ongoing evolution of the SARS-CoV-2 genome, researchers from the National Library of Medicine in Bethesda, Maryland, along with the Massachusetts Institute of Technology (MIT) in Cambridge, Massachusetts, constructed a phylogenetic tree of all available SARS-CoV-2 genomes. To this end, the researchers identified the sites within the SARS-CoV-2 genome that are subject to positive selection, which is a type of viral adaptation mechanism that is directly involved in virus-host coevolution.
“The fate of a novel zoonotic virus is, in part, determined by the race between public health intervention and virus diversification.”
Constructing an improved SARS-CoV-2 phylogenetic tree
In their work, the researchers constructed a global phylogenetic tree by collecting the genomes of all available SARS-CoV-2 sequences that had been identified prior to January 8, 2021. Taken together, a total of 321,096 sequences were submitted in the Gisaid database, of which 175,857 were found to exhibit unique SARS-CoV-2 genome sequences.
Further analysis of these genomes identified a total of 98,090 high-quality sequences, which were then used to construct the phylogenetic tree. Any mutations that were found to be repeated among the mutations in this tree were subsequently analyzed to identify genomic sites that were subject to positive selection.
Once the tree was constructed, eight principal partitions were identified, along with three divergent clades. Taken together, these partitions and clades can be identified by a specific amino acid replacement signature, which generally corresponded to the most prominent amino acid replacement across the tree.
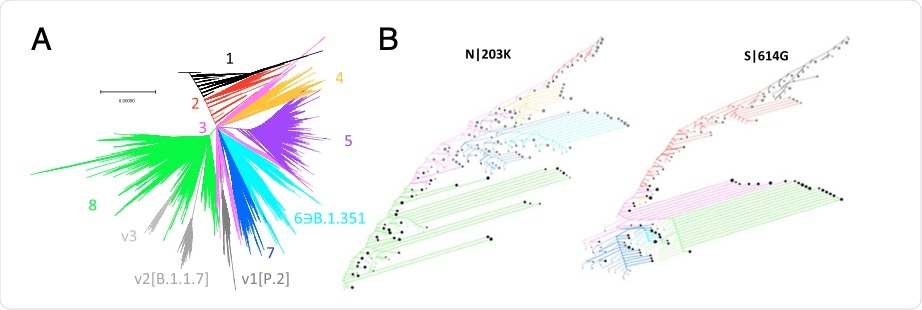 Global phylogeny of SARS-CoV-2. (A) Global tree reconstruction with eight principal partitions and three variant clades enumerated and color coded. (B) Site history trees for spike 614 and nucleocapsid 203 positions. Nodes were included in this reduced tree based on the following criteria: those immediately succeeding a substitution; those representing the last common ancestor of at least two substitutions; or terminal nodes representing branches of five se- quences or more (approximately, based on tree weight). Edges are colored according to their position in the main partitions, and the line type corresponds to the target mutation (solid) or any other state (dashed). Synonymous mutations are not shown. These sites are largely binary, as are most sites in the genome. The terminal node sizes are proportional to the log of the weight descendent from that node beyond which no substitutions in the site occurred. Node color corresponds to target mutation (black) or any other state (gray).
Global phylogeny of SARS-CoV-2. (A) Global tree reconstruction with eight principal partitions and three variant clades enumerated and color coded. (B) Site history trees for spike 614 and nucleocapsid 203 positions. Nodes were included in this reduced tree based on the following criteria: those immediately succeeding a substitution; those representing the last common ancestor of at least two substitutions; or terminal nodes representing branches of five se- quences or more (approximately, based on tree weight). Edges are colored according to their position in the main partitions, and the line type corresponds to the target mutation (solid) or any other state (dashed). Synonymous mutations are not shown. These sites are largely binary, as are most sites in the genome. The terminal node sizes are proportional to the log of the weight descendent from that node beyond which no substitutions in the site occurred. Node color corresponds to target mutation (black) or any other state (gray).
In these signature replacements, the RBD of the spike (S) protein, which is utilized by SARS-CoV-2 to gain entry into the host cell, as well as a region of the nucleocapsid (N) protein, were both associated with nuclear localization signals (NLS). Additionally, these NLS of the S and N proteins were also found in the nonstructural proteins of 1ab, 3a, and 8. These findings, therefore, suggest that positive selection plays a strong role in the evolution these specific areas of the SARS-CoV-2 genome.
Further examination of these mutations led to the discovery that both synonymous and nonsynonymous nucleic acid substitutions occurred among these variants. Out of all 12 possible nucleotide substitutions, C to U mutations were found to occur at a threefold greater frequency than any other nucleotide substitution, except for G to U substitutions.
The substantial purifying selection was detected across most of the SARS-CoV-2 genome, which agreed with previous studies that have found purifying selection to affect about 50% of the genomic sites in RNA viruses. Notably, the number of nonsynonymous substitutions significantly exceeded that of synonymous substitutions, which indicates ongoing and rapid host adaptation, as well as extremely high sampling density.
Assessing the presence of positive selection
Although the researchers confirmed that the evolution of SARS-CoV-2 is primarily due to purifying selection, they were interested in determining whether any of the nonsynonymous and repeated mutations could resolve to a single event. Positive selection was therefore deemed to be a plausible explanation for this observation.
In an effort to identify the sites that were likely to be subject to this form of selection, the researchers focused only on nonsynonymous substitutions that independently occurred at a frequency greater than 90% of all synonymous substitutions, aside from those that occurred in the NCN context.
The residues that were found to evolve under positive selection likely occurred due to a concurrence network that reflects epistatic interactions. More specifically, the researchers found that the central hubs of these interactions are the D614G in the S protein, as well as two adjacent substitutions of R203K and G204R in the N protein. Taken together, these three positively selected mutations were found to be most affected by this form of selection.
The significance of positive selection mutation
The positively selected D614G mutation of the S protein was found to likely increase the infectivity of SARS-CoV-2 by increasing the binding affinity between the S protein and the angiotensin-converting enzyme 2 (ACE2) receptor that resides on the surface of host cells. While the identification of this mutation has answered previous questions regarding extinct partitions, further information is still needed to determine whether this mutation is a passenger to another type of mutagenic or epidemiological event.
Another important finding in this study was that the D614G mutation appears to be a central hub for the epistatic network. This therefore indicates that any epistatic interactions that occur with this residue can lead to the establishment of mutations that can ultimately increase the viral fitness of a mutated progeny. Therefore, many, if not most, variants with the D614G mutation confer a substantial selective advantage.
Within the RBD, the N331 and N343 sites have been shown to be important for maintaining the infectivity of SARS-CoV-2. Therefore, substitutions like the N234Q, L452R, A475V, and V483A within the RBD have been found to confer some resistance to neutralizing antibodies. While these mutations did not meet the criteria in the current study to be determined as the results of positive selection, these mutations still appeared multiple times across the phylogenetic tree discussed here.
Study takeaways
In this study, the researchers obtained strong evidence of continuous virus diversification within geographic regions and ‘speciation,’ which was defined as the formation of stable, diverging, and region-specific variants. Overall, this ongoing adaptive diversification of SARS-CoV-2 could substantially prolong the pandemic and the vaccination campaign, in which variant-specific vaccines are likely to be required.
 How modern diets are driving rapid evolution in gut bacteria
How modern diets are driving rapid evolution in gut bacteria