The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that emerged in Wuhan, China, in December 2019, has infected over 237 million worldwide and caused the deaths of over 4.85 million. This has led to intensive research into the pathogenesis of the coronavirus disease 2019 (COVID-19) in order to control the clinical features and prevent disability and death.
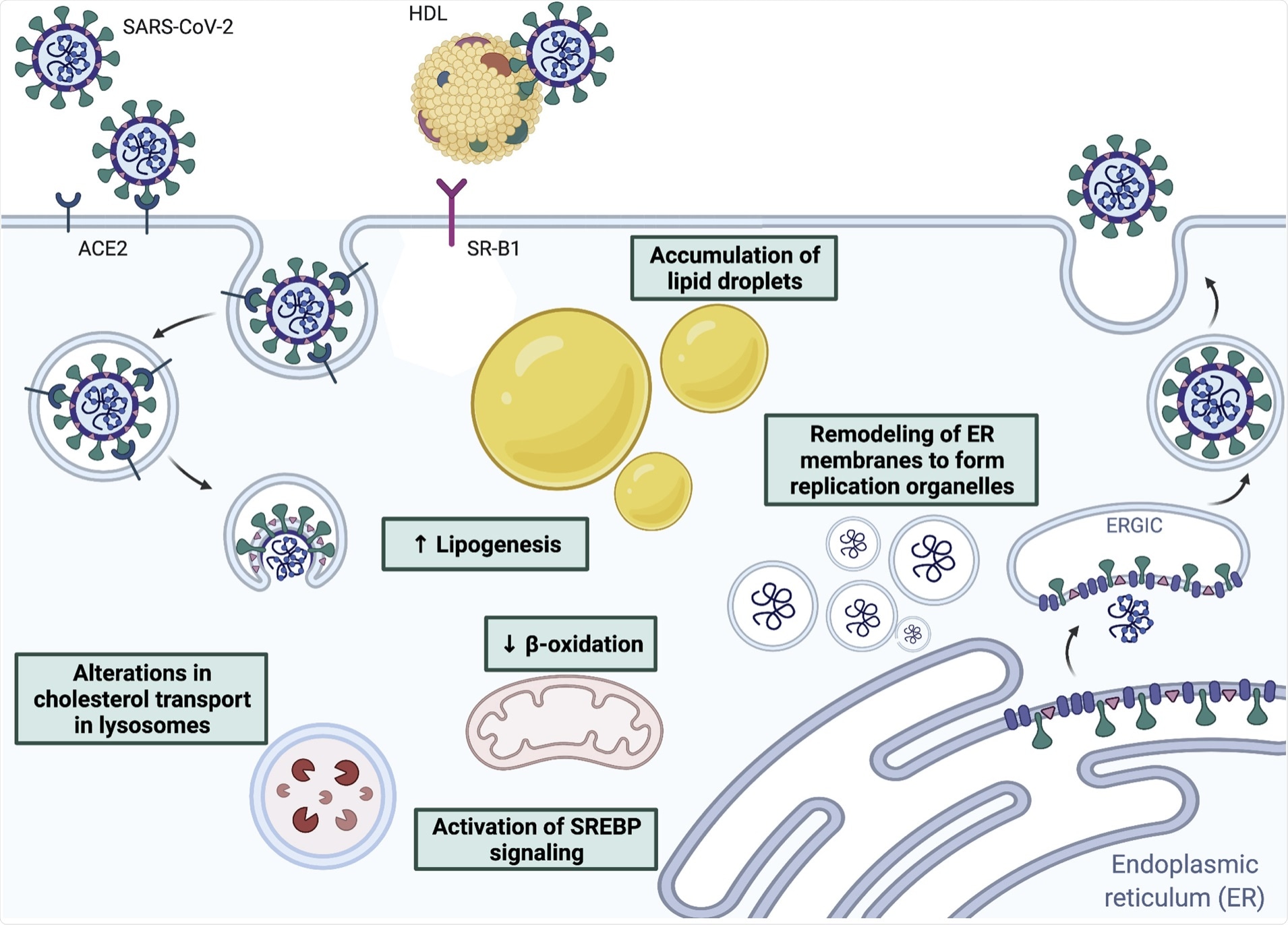 Interactions between coronaviruses and host lipids, including receptor binding and fusion, remodeling of endoplasmic reticulum-derived membranes to form replication organelles, and alterations in lipid metabolism to promote viral replication.
Interactions between coronaviruses and host lipids, including receptor binding and fusion, remodeling of endoplasmic reticulum-derived membranes to form replication organelles, and alterations in lipid metabolism to promote viral replication.
Background
Most viral respiratory illnesses in humans are caused by adenoviruses or by viruses with a ribonucleic acid (RNA) genome. Besides SARS-CoV-2, this includes respiratory syncytial virus (RSV), the influenza virus, the parainfluenza virus, and rhinoviruses.
These viruses mostly cause a mild upper respiratory infection. In severe cases, however, the lower respiratory tract is involved. Host immune responses are targeted at muting the clinical features of the infection, thus inducing disease tolerance or reducing the viral load by activating antiviral resistance. The outcome is to clear the infection.
The downside of the antiviral immune response is the adverse effects caused by the activation of pro-inflammatory mediators. These may trigger systemic hyper-inflammatory phenomena that cause severe tissue damage, resulting in multi-system dysfunction and acute respiratory distress syndrome (ARDS) that is characteristic of critically ill COVID-19 patients.
Host tolerance molecules are crucial to modulating this inflammatory signaling cascade and thus preventing severe harm to the host.
Lipids and viral entry
Viruses enter the host cell via their cell membranes, which are rich in lipids. During SARS-CoV-2 infection, the viral membrane attaches to the host cell membrane via fusion that is mediated by the viral spike protein following its binding to the host angiotensin-converting enzyme 2 (ACE2) cell receptor. The next step involves proteolytic cleavage of the spike at the interface between the two subunits.
The fusion step depends, however, on the addition of a palmitoyl group to the spike protein. Similarly, the spike protein must be acylated by the host ZDHHC20 enzyme for the virus to be infective, as this promotes spike-lipid membrane interactions.
This virus also binds cholesterol within high-density lipoprotein (HDL) particles. To this end, the HDL uptake by the HDL receptor scavenger receptor B type 1 (SR-B1) causes increased viral entry in ACE2-positive cells.
Multiple lipid molecules affect the fluidity and curvature of the membrane. For instance, phosphatidylethanolamine and cholesterol increase fluidity and negative membrane curvature, allowing viral fusion to occur. The opposite occurs with lysophospholipids.
Thus, compounds capable of altering the membrane lipid composition may inhibit infection by a number of viruses, increasing the spectrum of activity while reducing the possibility of resistance. The current study discusses the molecule LJ001, which is a photosensitizer that is activated by light to generate oxidative activity on unsaturated phospholipid substrates, thus making the membrane rigid and unable to participate in the viral fusion.
While membrane rigidity occurs in both host and viral membranes, only the former is capable of repair via the synthesis of new lipids. This compound may lead to the development of new types of antivirals through this mechanism of activity.
Other classes of potential antiviral compounds include rigid amphipathic fusion inhibitors (RAFIs) and nucleoside analogs that prevent negative curvature formation by incorporation into the membranes.
Lipid rafts are also important in facilitating viral endocytosis, as they are rich in cholesterol and glycosphingolipids, which express high levels of cell surface receptors. Acid and neutral sphingomyelinases can break sphingomyelin in lipid rafts down to ceramide, thus enhancing negative curvature and increasing fluidity.
Finally, lipids can alter the conformation of the receptors on the virus or host cells to inhibit viral binding and subsequent infection. The spike protein binds linoleic acid tightly, stabilizing it in the locked conformation so that it cannot engage with the ACE2 receptor. Omega-3 fatty acids, of which linoleic acid is a member, can thus inhibit viral infectivity.
Lipids and viral replication
Lipid molecules also hijack the cellular lipid pathways so as to ensure adequate oxidative substrates are available for viral replication processes. This occurs via the recruitment of phosphatidylinositol 4 (PI4)-kinase IIIβ in remodeling cell membranes.
Positive-sense RNA viruses use membrane-bound replication organelles (ROs) to increase the efficiency of viral replication and perhaps prevent the host antiviral response by masking the viral particles from immune recognition. For SARS-CoV-2, ROs come from the endoplasmic reticulum, mediated via viral non-structural proteins.
Cytosolic phospholipase A2α (cPLA2α) is also involved in this process and offers a target for reducing viral replication. Cholesterol biosynthesis is key to SARS-CoV-2 infection, including several genes that take part in the metabolism and transport of this lipid molecule.
Viral particles were also found to localize along with lipid droplets, which could mean that the latter offer a platform for replication. The SREBP pathway could thus be a therapeutic pan-coronavirus target.
In vitro screening of repurposed drugs shows that phospholipidosis is strongly linked to anti-SARS-CoV-2 activity. Further research is required to assess the clinical value of these findings.
Lipids and inflammation
Inflammatory mediators are also lipids that are derived from eicosanoids. These include prostanoids, comprising prostaglandins (PGs) and thromboxane (Tx), which are produced by cyclooxygenases (COXs)-1 and -2; leukotrienes via the lipoxygenase (LOX) enzymes; as well as epoxyeicosatrienoic acids (EETs) and 20-hydroxyeicosatetraenoic acid (20-HETE) that are formed by cytochrome P450 (CYP) enzymes.
While some of these derivatives are immunomodulators, they may also inhibit viral replication and the host immune response. Others could lead to a cytokine storm that triggers severe or critical COVID-19.
Mouse experiments indicate a favorable response to COX-2 inhibition, PGE2 inhibition, and PGD2 receptor DPr1 agonism or DPr2 inhibition. Other components of these pathways are also directly implicated in viral respiratory infections through their effect on host immunity or indirectly by promoting a fibrotic response.
Drugs like montelukast, which affects the LOX pathway, are suggested to be useful in hospitalized COVID-19 patients by reducing the risk of progression. However, clinical trials are still underway to assess their utility.
Epoxyeicosatrienoic acids (EETs) are powerful anti-inflammatory drugs that act by inhibiting cytokine-induced nuclear factor κB (NF-κB) activation and leukocyte adhesion to the vascular wall. 20-HETE has the opposite effect; however, their role in SARS-CoV-2-related immune responses is not yet clear.
Lipidomics in COVID-19
COVID-19 is associated with a shift to fatty acid oxidation, as shown in patients with trauma or acquired immunodeficiency syndrome (AIDS). Thus this is likely a common alteration in the metabolic response during a critical illness, and recovery is accompanied by the return of HDL and low-density lipoproteins (LDL) to the normal levels.
Sphingosine-1-phosphate (S1P) is reduced in COVID-19, perhaps because of the decreased HDL levels since the latter is the carrier molecule for S1P in blood. This reduction could lead to the dampening of many biological processes involved in inflammation and tissue damage.
Increased phospholipase A2 activity is also seen in COVID-19, as shown by the reduced glycerophospholipid and increased lysophospholipid levels. Eicosanoid synthesis is also increased, and elevated PLA2 levels may be an early signal of severe COVID-19.
However, only long-term follow-up will determine if this is true of lipid alterations in COVID-19 patients.
Conclusion
“The integral role of lipids in the viral life cycle suggests that targeting these pathways may be a viable therapeutic strategy. Serial lipidomic analyses in individuals with COVID-19 may identify specific lipid pathways that mediate the heterogenous response to viral infection, serve as prognostic biomarkers, or contribute to long-term sequelae.”
 APOE4 alters brain immunity and cognition differently in females and males
APOE4 alters brain immunity and cognition differently in females and males