Currently available treatments for coronavirus disease 2019 (COVID-19) are not very effective, so drug repurposing research efforts are needed to identify drug candidates that could be effective as COVID-19 treatments, particularly MPro inhibiting small molecules. In this context, boceprevir, an approved potent MPro inhibitor, has been explored extensively as a repurposed COVID-19 drug.
When SARS-CoV-2 infects human cells, its RNA genome translates into pp1a and pp1ab polypeptides, of which MPro is a protease/peptide fragment. These peptides undergo proteolytic hydrolysis to form non-structural proteins (nsps), which are essential for viral replication, evading the host immune system, and the packaging of new virions for infecting new host cells. The intervention of proteolytic hydrolysis is considered an effective approach to contain SARS-CoV-2 infection. As MPro processes the majority of nsps, it is the main target for broad-spectrum antiviral drug candidates against SARS-CoV-2.
 Study: The N-Terminal Carbamate is Key to High Cellular and Antiviral Potency for Boceprevir-Based SARS-CoV-2 Main Protease Inhibitors. Image Credit: NIAID
Study: The N-Terminal Carbamate is Key to High Cellular and Antiviral Potency for Boceprevir-Based SARS-CoV-2 Main Protease Inhibitors. Image Credit: NIAID
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
The study
The researchers designed and synthesized a total of 19 boceprevir-based MPro inhibitors from MPI29 to MP147, and one PF07321332 inhibitor, a Pfizer compound included due to its similarity with boceprevir.
Boceprevir-based MPro inhibitors were synthesized by introducing modifications at all of its four binding pockets - α ketoamide warhead, a P1 b-cyclobutylalanyl moiety, a P2 dimethyl-cyclopropyl proline, a P3 tert-butylglycine, and a P4 N-terminal tert-butylcarbamide.
The researchers followed the guidelines of peptide coupling while synthesizing these inhibitors and characterized in vitro MPro inhibition potency by determining their IC50 values following an established protocol that used a fluorogenic substrate Sub3. They also characterized the crystal structures of MPro bound with ten inhibitors using X-ray crystallography and antiviral potency of inhibitors in vitro and in human host cells (in cellulo).
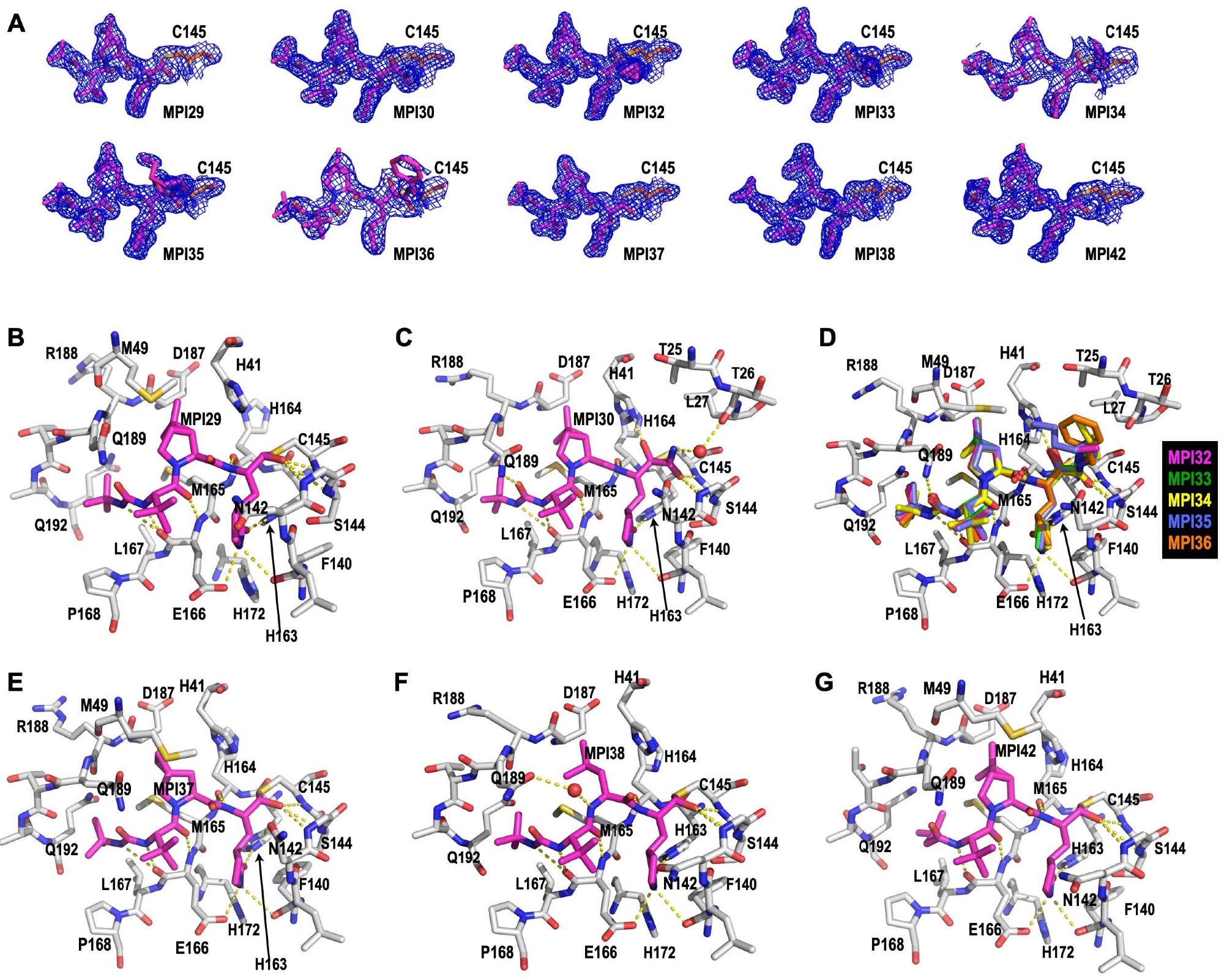 Crystal structures of MPro bound with 10 MPIs. (A) Contoured 2Fo-Fc maps at the 1" level around 10 MPIs and C145 in the active site of MPro. The active site structures for MPro bound with (B) MPI29, (C) MPI30, (D) MPI32-36, (E) MPI37, (F) MPI38, and (G) MPI42. Dashed yellow lines between inhibitors and MPro are potential hydrogen bonds.
Crystal structures of MPro bound with 10 MPIs. (A) Contoured 2Fo-Fc maps at the 1" level around 10 MPIs and C145 in the active site of MPro. The active site structures for MPro bound with (B) MPI29, (C) MPI30, (D) MPI32-36, (E) MPI37, (F) MPI38, and (G) MPI42. Dashed yellow lines between inhibitors and MPro are potential hydrogen bonds.
Results
The inhibition curves of MPro inhibitors were well defined, showed 100% antiviral activity without an inhibitor, and reached ~100% viral inhibition at 10µM inhibitor concentrations.
Of all the inhibitors, while MPI29 showed the highest in vitro potency and had an IC50 value of 9.3 nM, MPI30 showed lower in vitro potency, indicating the importance of the P1 opal residue in improving interactions with MPro. MP130 was also 100-fold more potent than boceprevir and had an IC50 value of 40 nM. Although both MPI29 and MPI30 showed very low in cellulo potency, they were highly potent MPro inhibitors in vitro.
All inhibitors from MPI31-36 showed much higher in vitro IC50 values than MPI30 due to an additional chemical appendage in the α-ketoamide, as nitrogen leads to less favored interactions with MPro. Except for MPI45, all other inhibitors with a P4 N-terminal carbamide cap displayed low cellular and antiviral potency. Although structurally similar to MPI47, PF-07321332 showed a 10-fold lower IC50 value than MPI47 due to a P4 N-terminal trifluoroacetamide cap responsible for its unique interactions with MPro. The inhibitors having the P1 opal residue, P2 dimethyl-cyclopropyl proline, and P4 N-terminal tert-butylcarbamide moieties made stronger hydrophobic interactions with MPro, subsequently showing high in vitro potency.
Furthermore, the crystal structure analysis showed that all the inhibitors formed a covalent adduct with the MPro active site cysteine. In its MPro complex structure, an inhibitor containing a P4 N-terminal isovaleramide was tucked deep in a small pocket recognized as a P4 alanine side chain in a substrate.
Although all the inhibitors showed high in vitro potency, their in cellulo potency was substantially different in human 293T cells. While the inhibitors with a P4 N-terminal carbamide or amide showed low in cellulo potency, changing the P4 N-terminal cap to a carbamate reversed this trend. Similarly, introducing a P3 O-tert-butyl-threonine improved in cellulo potency.
Conclusions
Overall, the study results suggest that replacing the α ketoamide warhead with an aldehyde and P1 site with a b-(S-2-oxopyrrolidin3-yl)-alanyl (opal) residue leads to high in vitro potency. Also, the original boceprevir moieties at P2, P3, and the P4 N-terminal positions showed higher in vitro potency than other chemical moieties tested.
Based on the study findings, although boceprevir inhibits MPro, its inhibiting potency in a human cell (in cellulo) is weak, making it moderately effective against SARS-CoV-2. However, all the three boceprevir-derived inhibitors with a P4 N-terminal carbamate show high cellular potency and also high antiviral potency against SARS-CoV-2.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
The N-Terminal Carbamate is Key to High Cellular and Antiviral Potency for Boceprevir-Based SARS-CoV-2 Main Protease Inhibitors, Yugendar R. Alugubelli, Zhi Zachary Geng, Kai Yang, Namir Shaabani, Kaustav Khatua, Xinyu R. Ma, Erol C. Vatansever, Chia-Chuan Cho, Yuying Ma, Lauren Blankenship, Ge Yu, Banumathi Sankaran, Pingwei Li, Robert Allen, Henry Ji, Shiqing Xu, Wenshe Ray Liu, bioRxiv (2021), 12.18.473330 doi: https://doi.org/10.1101/2021.12.18.473330, https://www.biorxiv.org/content/10.1101/2021.12.18.473330v1
- Peer reviewed and published scientific report.
Alugubelli, Y. R., Geng, Z. Z., Yang, K. S., Shaabani, N., Khatua, K., Ma, X. R., Vatansever, E. C., Cho, C.-C., Ma, Y., Xiao, J., Blankenship, L. R., Yu, G., Sankaran, B., Li, P., Allen, R., Ji, H., Xu, S., & Liu, W. R. (2022). A systematic exploration of boceprevir-based main protease inhibitors as SARS-CoV-2 antivirals. European Journal of Medicinal Chemistry, 240, 114596. https://doi.org/10.1016/j.ejmech.2022.114596. https://www.sciencedirect.com/science/article/pii/S0223523422004986.
 Long COVID symptoms change for 15 months after infection
Long COVID symptoms change for 15 months after infection