 *Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Background
The ongoing coronavirus disease 2019 (COVID-19) pandemic has led to ongoing global research efforts to understand the mechanisms of adaptation of SARS-CoV-2. Consequently, researchers and medical professionals have contributed significantly by sequencing the genome of SARS-CoV-2 from patient isolates at an unprecedented speed and publishing their findings on the Global Initiative on Sharing All Influenza Data (GISAID) database. It is important to understand the pathogenic mechanisms to successfully develop drugs, as well as establish effective health surveillance measures and immunological interventions.
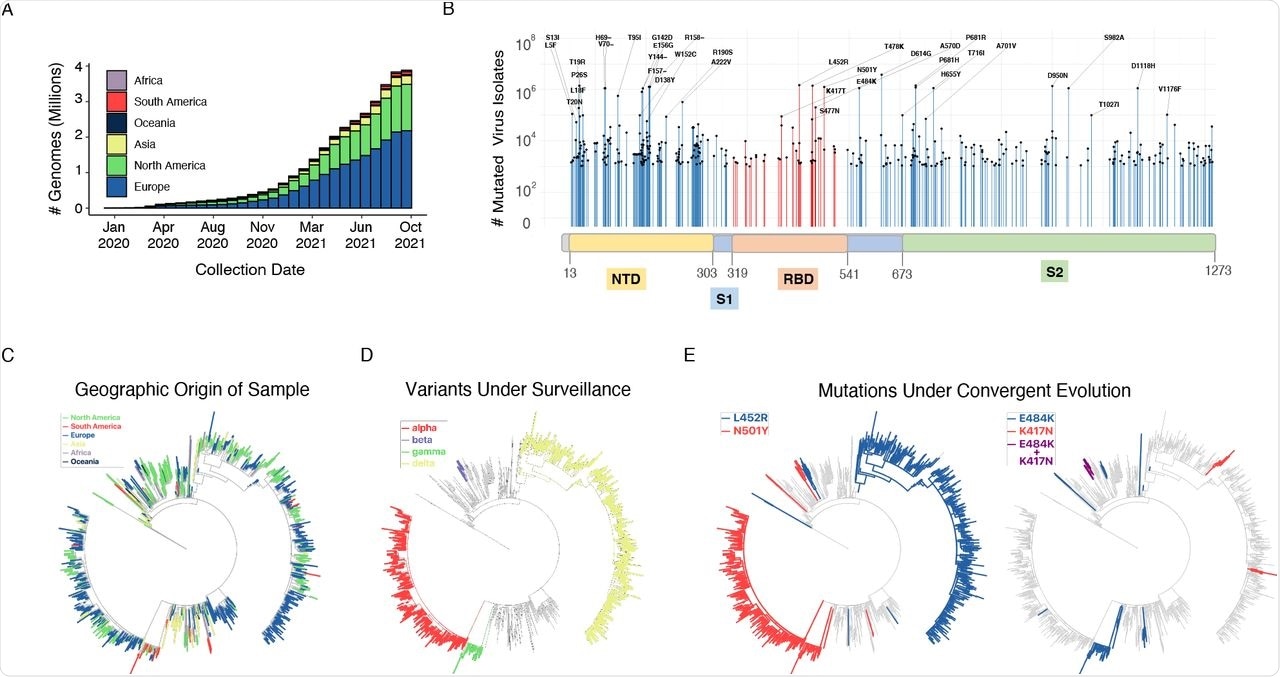
(A) Cumulative count of SARS-CoV-2 genome sequences in GISAID by collection date and the continent of sample origin as of September 26, 2021. (B) Distribution of high-frequency variants in the spike protein sequence. The RBD and NTD domains are highlighted orange and yellow, respectively. For a number of residues with high deviation from wild type sequence (417, 452, 484, 501, 614), the most common mutation is indicated. (C) SARS-CoV-2 maximum-likelihood phylogeny of a high-diversity 1000-genome subset. The tree is an outgroup rooted with the related bat CoV genome RATG13. The root branch length is truncated to emphasize relationships amongst SARS-CoV-2 genomes. The geographic origin of each of the sequences is denoted by branch color. (D) Distribution of haplotypes of the 4 variants of concern (alpha, beta, gamma, delta), mapped onto the phylogeny. (E) Distribution of mutations under convergent evolution (L452R, E484K), mapped onto the phylogeny.
Despite global efforts, there is a limited understanding as to how SARS-CoV-2 maintains the conservation and diversity of its genome. New mutations continue to arise spontaneously in the viral genome, thus leading to the emergence of new variants or the loss of variants, which could benefit the virus within or across host environments and diversify or conserve the viral protein domains. This genetic adaptation is key to the successful transmission of SARS-CoV-2 in humans.
The selective force driving the conservation of amino acids in viruses is known as purifying or negative selection, which helps protect the integrity of a domain structure or function. In contrast, positive selection is the accumulation of amino acid changes, which might be due to the gain of function through molecular alterations.
The tendency to escape host-immune factors, such as antibodies and T-cells, often leads to a positive selection of viral antigens. Antigenic variation can affect current therapeutics and vaccine efficacy, as well as cause re-infections or persistent viral infections. Therefore, it is crucial to characterize the conserved and diverse regions of the viral genomes while emphasizing the antigenic targets to develop therapeutics and understand drug resistance.
Study findings
The authors compared the identity of coding sequences from about four million curated sequences, of which about 41% were found in a single isolate (singleton). The team reported that at least 2,000 high-frequency variant (HFV) isolates and shared 1,689 amino acid substitutions. The team observed that within the spike protein, HFVs were accumulated in the N-terminal domain (NTD).
Convergent evolution of mutations was identified by comparing the diversity of individual genomes based on lineages. Mutations like L452R and E484K were detected independently in genomes from at least five and seven lineages, respectively.
Further, the mutation rate for the SARS-CoV-2 genome was calculated to be about 7 x 10-4 nucleotide substitutions/site/year based on current Bayesian estimates, which is lower than other human coronaviruses and other ribonucleic acid (RNA) viruses. This might suggest that purifying or negative selection is dominant in the SARS-CoV-2 genome.
The researchers employed FUBAR, a Bayesian codon model, to find sites that evolved by natural selection. To this end, the SARS-CoV-2 genome appears to be strongly under negative/purifying selection; however, many sites were identified in several genes under positive selection.
Substitutions at sites in the receptor-binding domain (RBD) due to purifying selection reported a maximal negative impact on angiotensin-converting enzyme 2 (ACE2) receptor binding affinity and protein stability. Conversely, positive selection in the RBD reported less or no effect on ACE2 binding and protein stability. The researchers also observed that T-cell epitope sequences were highly conserved due to strong purifying selection.
Conclusions
The present study made a significant observation that SARS-CoV-2 antigen and epitope conservation is critical. Furthermore, the researchers noted that the SARS-CoV-2 RBD mediates entry of the virus into host cells and is also the prime target of neutralizing antibodies.
The spike protein was also found to lack any enrichment of sequence diversity in comparison to the whole SARS-CoV-2 genome. Genes like N, ORF3a, ORF7a, ORF8a, and ORF10 reportedly contained several sites under positive selection that are indicative of distinct selection. Further, the authors observed that most of these positively selected genes were responsible for the suppression of innate immune responses.
In conclusion, the current study reports that the SARS-CoV-2 genome, as a whole, is strongly under a purifying selective force, with a minor number of sites being positively selected due to a diversifying force. These sites are critical for protein stability and reduced neutralization by convalescent sera. Based on their observations, the authors proposed that SARS-CoV-2 genetic selection is a naturally purifying selection by a deterministic process.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
 RSV and COVID-19 trigger different immune responses in infants
RSV and COVID-19 trigger different immune responses in infants