The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which has led to the current coronavirus disease 2019 (COVID-19) pandemic, is an enveloped ribonucleic acid (RNA) virus that belongs to the genus Betacoronavirus. The Betacoronavirus genus also comprises SARS-CoV-1, which led to the 2003 SARS outbreak, as well as the Middle East respiratory syndrome coronavirus (MERS-CoV), which led to the 2012 MERS outbreak.
Despite the devastating impact of SARS-CoV-2 on public health and the global economy, the distribution of COVID-19 vaccines around the world remains challenging. Furthermore, the first two therapeutics that can significantly reduce mortality associated with COVID-19 were not identified until late 2021. Therefore, knowledge of the unique RNA biology of SARS-CoV-2 is important for the development of new therapeutics against this virus, as well as other Betacoronaviruses.
SARS-CoV-2 is the largest known RNA virus whose genome consists of positive-sense single-stranded RNA (ssRNA). Previous studies on the secondary structure of the coronavirus genome revealed the 5′ untranslated region (UTR), the 3′ UTR, and the frameshifting stimulation element (FSE) conserved regions essential for viral replication.
The role of SARS-CoV-1 and SARS-CoV-2 FSEs
Approximately the first two-thirds of the coronavirus genome consists of one open reading frame (ORF1) that encodes 16 non-structural proteins (nsps). ORF1 is partitioned into an upstream ORF1a and a downstream ORF1b by a stop codon that is located in the middle of ORF1.
Although some ribosomes stop after translation of polyprotein ORF1a, the frameshifting stimulation element (FSE) causes few ribosomes to slip backward by one nucleotide and bypass the stop codon, thereby translating the entire ORF1ab.
Several ORF1ab proteins were found to be essential for RNA replication and transcription. Furthermore, many studies have indicated that an optimal ribosomal frameshifting rate is critical.
Any small difference in the percentage of frameshifting can bring about significant differences in genomic RNA production and infectivity. Therefore, FSE can be considered a major drug target for small molecules and needs to be investigated for its role in the treatment of SARS-CoV-2.
FSEs from both SARS-CoV-1 and SARS-CoV-2 have been found to fold into a complex structure with a three stemmed pseudoknot. Despite the importance of the FSE structure, no information regarding the relationship between the RNA folding conformation and frameshifting rate in infected cells is available.
Background
Recent advances in RNA chemical probing have enabled genome-wide characterization of RNA structures that are present in living cells. Dimethyl sulfate (DMS) and reagents in the SHAPE and icSHAPE families are the most commonly used chemical probes.
Prediction of RNA structures is more accurate with DMS as compared to SHAPE. However, the RNA genomes of viruses form many structures that cannot be determined accurately by chemical probes. Therefore, more work is required to determine the dynamics of the RNA structures within the SARS-CoV-2 genome, as well as their functional roles.
A new study published in Nature Communications performed DMS mutational profiling with sequencing (DMS-MaPseq) and DREEM clustering using infected Huh7 and Vero cells for the determination of the SARS-CoV-2 RNA secondary structure.
About the study
The current study involved infection of monkey Vero cells and human Huh7 cells with SARS-CoV-2. Thereafter, these cells underwent DMS modification followed by RNA extraction and ribosomal RNA (rRNA) subtraction. Following rRNA subtraction, the DMS-modified RNA was used for the generation of the DMS-MaPseq library.
In vitro, FSE transcription and DMS modification were carried out followed by ex-virion RNA extraction and DMS modification. Dual-luciferase frameshift reporter assay was used to determine frameshift efficiency.
A bit vector, which was of the length of the reference sequence, was generated using DREEM to map and quantify mutations in the SARS-CoV-2 genome. Few of the bit vectors were filtered if they had more than an allowed total number of mutations, had two mutations closer than four nucleotides apart, or had a mutation next to an uninformative bit.
Genome-wide coverage was computed with the help of unfiltered bit-vectors and DMS/SHAPE reactivity correlations were computed using filtered bit vectors. Thereafter, the entire SARS-CoV-2 genome was folded based on DMS activities from Vero and Huh7 cells. The area under the receiver operating characteristic curve (AUROC) computation was done to determine how well DMS/SHAPE reactivities support the predicted RNA structure.
Similarities between the two RNA structures were determined with the help of the modified Fowlkes-Mallows index (mFMI). For decoy structures, AUROC was computed based on previously collected DMS-MaPseq data.
Following this, FSE folding was carried out from Vero and Huh7 cell data. Coronavirus sequence alignment was also conducted, followed by the detection of alternative structures within the Vero cells.
Covariation among paired bases in the SARS-CoV-2 genome structure was analyzed. Finally, the negative strands were quantified and RNA structures were visualized.
Study findings
The DMS reactivities of SARS-CoV-2 were found to be similar in both Vero and Huh7 cells. The AUROC values from Huh7 and Vero cells were found to be 0.99 and 0.98, respectively, which indicated that the in-cell data was of high quality. Five stem-loops (SL1–5) were also found within the 5′ UTR, and three stem-loops (SL6–8) were found downstream of 5′ UTR.
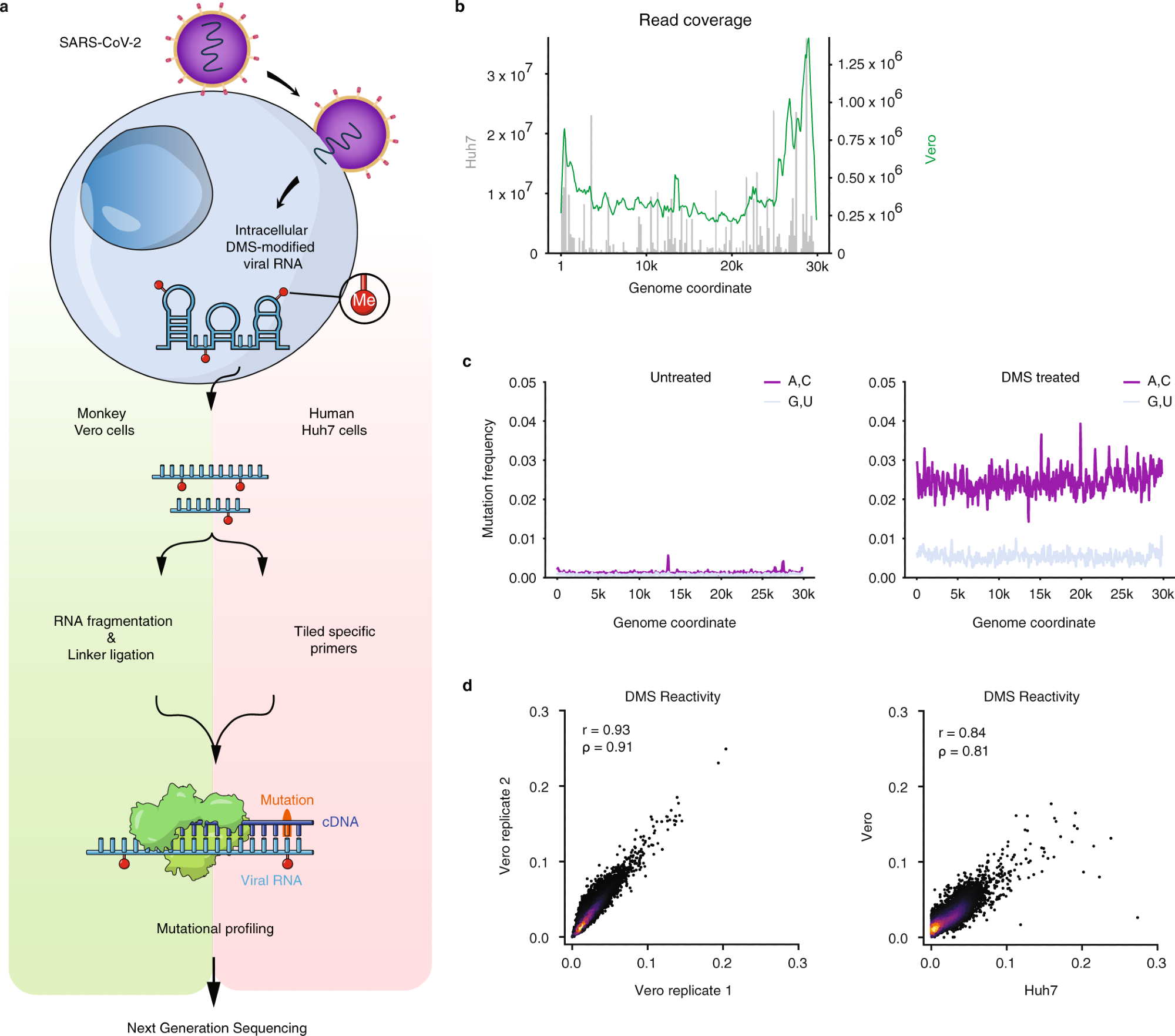 a Schematic of the experimental protocol for probing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA structures in Vero and Huh7 cells using dimethyl sulfate mutational profiling with sequencing (DMS-MaPseq). b Read coverage as a function of genome coordinate for Huh7 cells using tiling specific primers (gray bars, left axis) and Vero cells using linker ligation (green curve, right axis); Vero coverage was smoothed by taking the mean over a sliding window of 500 nt. c Signal vs. noise plots of mutation frequencies (i.e., among all reads aligning to each genome coordinate, the fraction of reads with a mutation at that coordinate) on adenines (As) and cytosines (Cs) vs. guanines (Gs) and uracils (Us) as a function of genome coordinate for untreated and DMS-treated RNA. A mutation frequency of 0.01 at a given position represents 1% of reads having a mismatch or deletion at that position. Signal and noise were smoothed by taking the mean over 100 nt windows in increments of 50 nt. d Comparison of DMS reactivities on As and Cs between biological replicates in Vero cells (left) and between the averaged of Vero replicates and Huh7 cells (right). Pearson (r) and Spearman (ρ) correlation coefficients are shown. For each sample, the top 0.05% of mutational fractions (values over 0.27 for Vero and 0.38 for Huh7) were considered outliers and excluded from the plot and calculation of correlation coefficients.
a Schematic of the experimental protocol for probing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA structures in Vero and Huh7 cells using dimethyl sulfate mutational profiling with sequencing (DMS-MaPseq). b Read coverage as a function of genome coordinate for Huh7 cells using tiling specific primers (gray bars, left axis) and Vero cells using linker ligation (green curve, right axis); Vero coverage was smoothed by taking the mean over a sliding window of 500 nt. c Signal vs. noise plots of mutation frequencies (i.e., among all reads aligning to each genome coordinate, the fraction of reads with a mutation at that coordinate) on adenines (As) and cytosines (Cs) vs. guanines (Gs) and uracils (Us) as a function of genome coordinate for untreated and DMS-treated RNA. A mutation frequency of 0.01 at a given position represents 1% of reads having a mismatch or deletion at that position. Signal and noise were smoothed by taking the mean over 100 nt windows in increments of 50 nt. d Comparison of DMS reactivities on As and Cs between biological replicates in Vero cells (left) and between the averaged of Vero replicates and Huh7 cells (right). Pearson (r) and Spearman (ρ) correlation coefficients are shown. For each sample, the top 0.05% of mutational fractions (values over 0.27 for Vero and 0.38 for Huh7) were considered outliers and excluded from the plot and calculation of correlation coefficients.
A total of 95 base pairs were supported by covariation. The elements with the most covarying pair were found to be SL8 downstream of the 5’ UTR (two pairs), a short, unannotated hairpin near the 5′ end of the N gene (five pairs), and the stem containing s2m in the 3′ UTR (four pairs).
The majority of the SARS-CoV-2 genome was also found to form alternative structures. Decoy structures that were similar to the true structures were reported to have high AUROC, while those different from the true structures had low AUROC. FSEs also formed at least two distinct structures in both Vero and Huh7 cells.
The presence of Alternative Stem 1 (AS1) was also identified as the predominant FSE structure, rather than the three-stemmed pseudoknot. Furthermore, the AS1 pairing sequence was found to be conserved in all 12 of the SARS-related viruses, including SARS-CoV-1 and six other viruses that were isolated from bats. The FSE was also reported to fold properly in the absence of protein factors.
Analysis of intracellular folding using DREEM indicated the presence of at least two distinct conformations of FSEs in both Vero and Huh7 cells. The frameshifting rate of the long FSE was approximately 42%, while for the short FSE it was approximately 17%.
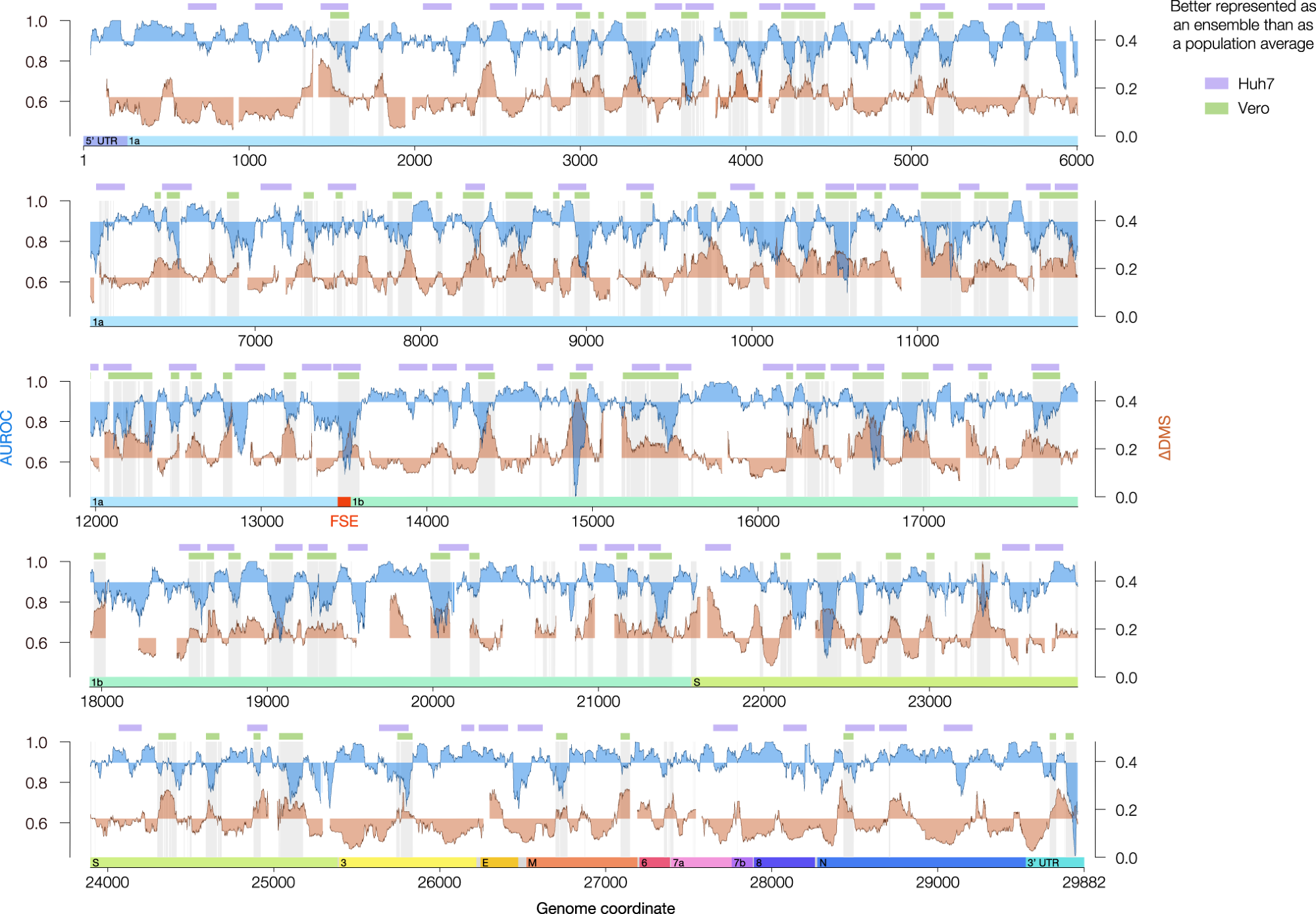 Agreement between DMS reactivities and predicted secondary structures (AUROC, blue) and the difference in DMS reactivity between clusters 1 and 2 (∆DMS, orange) for the genome-wide model in Vero. Both quantities were calculated over sliding windows of 80 nt in 1 nt increments; x values represent the centers of the windows. Windows with <10 paired or <10 unpaired bases were excluded from the calculation of AUROC; windows with <10 bases that clustered into at least two structures were excluded from the calculation of ∆DMS. For AUROC and ∆DMS, the area between the local value and the genome-wide median is shaded. For the Vero model, all coordinates best described by structure ensembles (AUROC below median, ∆DMS above median) are shaded in light gray. The green bars represent a denoised version of these coordinates (see Methods). For the Huh7 model, regions meeting criteria for alternative structures (see Methods) are labeled with lavender bars. The locations of the untranslated regions (UTRs) and open reading frames (ORFs) of SARS-CoV-2 are indicated below the AUROC and ∆DMS data. The frameshifting stimulation element (FSE, coordinates 13,462–13,546) is highlighted in red. Source data are provided as a Source Data file.
Agreement between DMS reactivities and predicted secondary structures (AUROC, blue) and the difference in DMS reactivity between clusters 1 and 2 (∆DMS, orange) for the genome-wide model in Vero. Both quantities were calculated over sliding windows of 80 nt in 1 nt increments; x values represent the centers of the windows. Windows with <10 paired or <10 unpaired bases were excluded from the calculation of AUROC; windows with <10 bases that clustered into at least two structures were excluded from the calculation of ∆DMS. For AUROC and ∆DMS, the area between the local value and the genome-wide median is shaded. For the Vero model, all coordinates best described by structure ensembles (AUROC below median, ∆DMS above median) are shaded in light gray. The green bars represent a denoised version of these coordinates (see Methods). For the Huh7 model, regions meeting criteria for alternative structures (see Methods) are labeled with lavender bars. The locations of the untranslated regions (UTRs) and open reading frames (ORFs) of SARS-CoV-2 are indicated below the AUROC and ∆DMS data. The frameshifting stimulation element (FSE, coordinates 13,462–13,546) is highlighted in red. Source data are provided as a Source Data file.
Conclusions
The current study provides significant insights on major RNA structures and sites of RNA structure heterogeneity across the entire SARS-CoV-2 genome. Furthermore, the researchers revealed that small molecules and/or antisense oligos can be designed to abolish SARS-CoV-2 frameshifting and can therefore be used as therapeutics.
Further work must be conducted to determine other structured elements across the SARS-CoV-2 genome that will help in the design of more targeted therapeutics.
Journal reference:
- Lan, T. C. T., Allan, M. F., Malsick, L. E., et al. (2022). Secondary structural ensembles of the SARS-CoV2 RNA genome in infected cells. Nature Communications. doi:10.1038/s41467-022-28603-2.
 11 genetic loci that shape impulsive decision-making
11 genetic loci that shape impulsive decision-making