This clustering tool utilizes sequence data collected over time to produce more stable clusters than other commonly used phylogenetic clustering methods. Moreover, their method is provided as an R package, thereby allowing for its use within research and public health community settings to investigate transmission dynamics of SARS-CoV-2.

Study: Cov2clusters: genomic clustering of SARS-CoV-2 sequences. Image Credit: Coffeemill / Shutterstock.com
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Background
The rapid development of coronavirus disease 2019 (COVID-19) vaccines, in addition to the implementation of non-pharmaceutical/social distancing measures, has successfully alleviated the impact of the pandemic by reducing viral transmission, hospitalization, and mortality rates. Nevertheless, COVID-19 remains a worldwide concern due to the continued emergence of more transmissible and virulent SARS-CoV-2 variants of concern (VOCs), waning vaccine-induced antibodies, vaccine hesitancy, and unequal access to vaccines and therapeutics.
An increasing amount of SARS-CoV-2 whole genome sequence (WGS) data is being shared every day through global repositories, which allows almost real-time genomic comparison of the pathogen. These data can be utilized to develop novel and easy-to-implement tools that can identify clusters of linked cases aiding in the understanding of regional epidemiology and informing public health policies, such as implementing restrictions in certain settings with a high transmission risk.
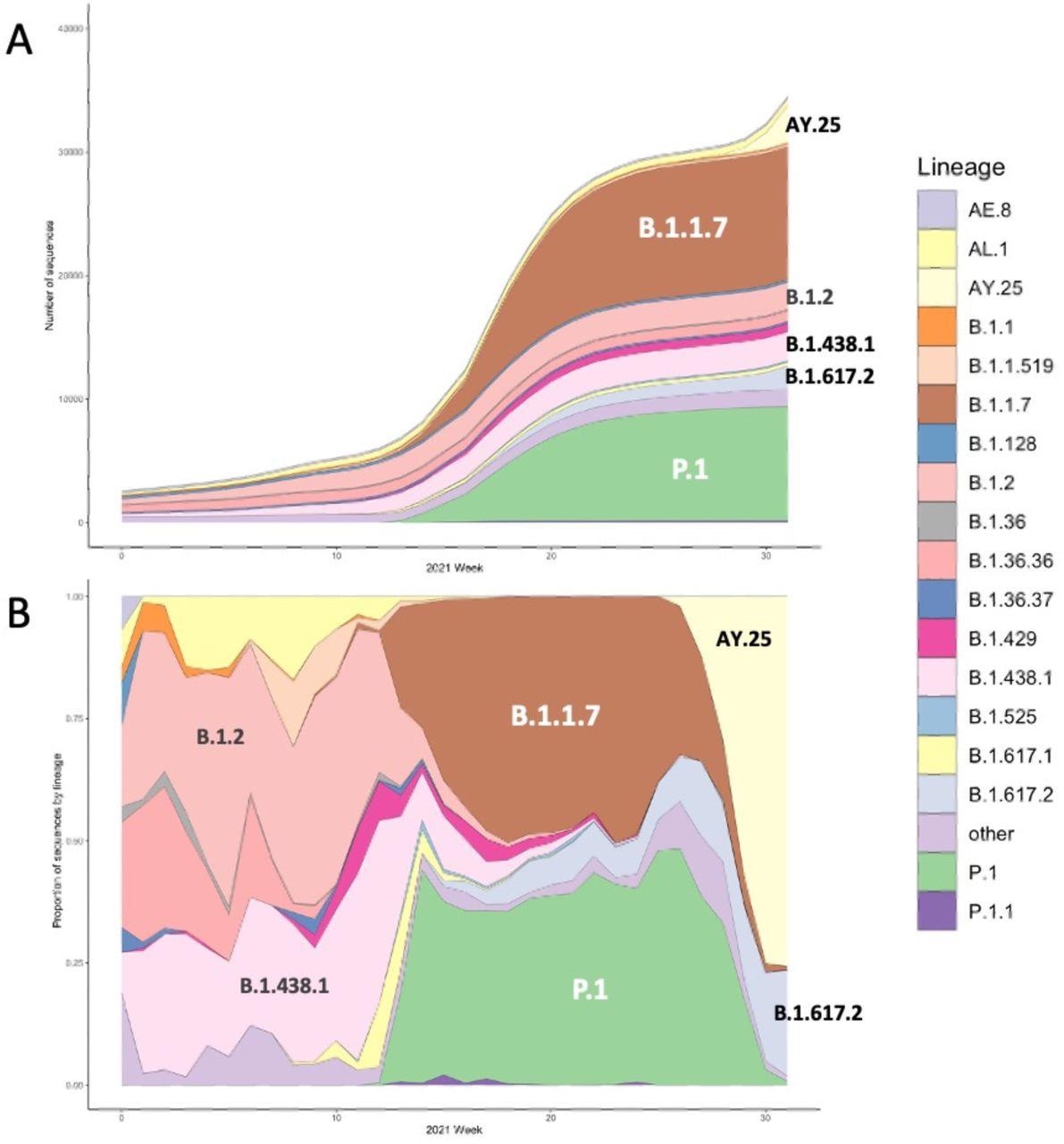
The cumulative number (A) and lineage proportion (B) of SARS-CoV-2 sequences per week included in the study, coloured by lineage. Major lineages present in the data are annotated.
The utility of defining SARS-CoV-2 clusters
Genomically-linked cases with shared demography should be identified at a higher resolution than a shared lineage assignment or simply through contact tracing. Currently, the Pangolin system is used for assigning nomenclature to SARS-CoV-2 lineages; however, Pangolin has been dynamic through the pandemic and cannot provide sufficient resolution for epidemiological investigations.
Thus, the researchers of the current study recommend a system where the clustering of sequences by genomic similarity is aided by epidemiological information. This would consequently provide a resolution and stability that is necessary for public health applications over the course of a dynamic pandemic.
To date, phylogenetic tree clustering methods have been applied to identify putative transmission clusters in SARS-CoV-2 based on genomic divergences. However, due to the rapid spread of the SARS-CoV-2 with relatively lesser alterations in genetic diversity, as well as periods of lineage replacement with new VOCs with reduced regional genetic diversity in the virus, clustering-based solely on genetic variation may not be sufficient to effectively identify meaningful clusters in SARS-CoV-2. Moreover, defining clusters using a fixed genetic distance threshold may cause sequences to alter cluster designation over time as more sequences become available.
Improved resolution and sensitivity of Cov2clusters
Through the use of their novel method to construct SARS-CoV-2 genomic clusters, the researchers use the pairwise probability of clustering under a logit regression model, wherein they link cases under a given probability threshold. The model uses a logit regression model based on sequence divergence and the sample collection dates. The model is flexible enough to add further resolution to this clustering by incorporating epidemiological data, such as geography, contact data, and exposure events.
In contrast to previous clustering approaches that often rely solely on phylogenetic inference (tree cluster reference), clustering isolates in this pairwise manner allows for greater cluster stability through time, as well as resolution by including epidemiological information without the need for time-consuming manual investigation.”
The team tested their novel method on SARS-CoV-2 sequence data collected during the first, second, and third waves of the COVID-19 pandemic in the British Columbia province of Canada from March 15, 2020, to August 13, 2021.
The results of the novel genomic clustering method were compared at three pairwise probability thresholds of 0.7, 0.8, and 0.9 for linking sequences to form clusters. To this end, the researchers found that their approach formed the most stable clusters at a probability threshold of 0.8 in the clinical data.
When compared to other phylogenetic clustering tools, the sensitivity of Cov2clusters at a 0.8 probability threshold was higher than both TreeCluster ‘max_clade’ and ‘single_linkage.” Furthermore, the produced clusters were more stable as cases were added over time.
This result has particular significance for the utility of this method in real-time public health surveillance, where sequencing datasets grow over time, and stability in cluster designations is beneficial for reporting and surveillance.”
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Sobkowiak B., Kamelian, K., Zlosnik, J. E. A., et al. (2022) Cov2clusters: genomic clustering of SARS-CoV-2 sequences. medRxiv. doi:10.1101/2022.03.10.22272213, https://www.medrxiv.org/content/10.1101/2022.03.10.22272213v2
- Peer reviewed and published scientific report.
Sobkowiak, Benjamin, Kimia Kamelian, James E. A. Zlosnik, John Tyson, Anders Gonçalves da Silva, Linda M. N. Hoang, Natalie Prystajecky, and Caroline Colijn. 2022. “Cov2clusters: Genomic Clustering of SARS-CoV-2 Sequences.” BMC Genomics 23 (1). https://doi.org/10.1186/s12864-022-08936-4. https://bmcgenomics.biomedcentral.com/articles/10.1186/s12864-022-08936-4.
 COVID-19 immunity stays specific and barely boosts protection against common cold coronaviruses
COVID-19 immunity stays specific and barely boosts protection against common cold coronaviruses