Since its emergence at the end of 2019, the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has infected over 526 million worldwide and caused over 6.28 million deaths. SARS-CoV-2 primarily infects the human respiratory epithelial cells by binding to the cell surface angiotensin-converting enzyme 2 (ACE2) receptor.
The receptor-binding domain (RBD) of the SARS-CoV-2 spike (S) protein interacts with ACE2 to mediate entry of the virus into the host cell. The SARS-CoV-2 S protein consists of two segments including S1, which contains the RBD, and S2, which mediates host and viral membrane fusion.
Background
Fast and accurate diagnostic tools, such as the reverse-transcription polymerase chain reaction (RT-PCR) assay and coronavirus disease 2019 (COVID-19) rapid antigen test kits, along with necessary quarantine and medical measures, are important for preventing SARS-CoV-2 transmission. Most COVID-19 rapid antigen test kits depend on specific antigen-antibody reactions and have a sensitivity of approximately 85%.
DNA or ribonucleic acid (RNA) aptamers are short and single-stranded (ss) nucleotides that can be used instead of antibodies in immunoassays. Aptamers have several advantages as compared to antibodies such as low cost, reusability, robustness, can be easily modified chemically, and low immunogenicity.
Aptamers that target both the SARS-CoV-2 RBD and nucleocapsid (N) protein have been studied to determine their potential use as antiviral therapy. Several studies have also employed in silico approaches to screen or design aptamers that target the SARS-CoV-2 S protein.
A new International Journal of Molecular Sciences study used the known sequence of SARS-CoV-2 against aptamers to perform the in silico selection of an aptamer against the SARS-CoV-2 S protein.
Study: In-Silico Selection of Aptamer Targeting SARS-CoV-2 Spike Protein. Image Credit: Cinefootage Visuals / Shutterstock.com
About the study
The current study involved the acquisition of the SARS-CoV-2 S protein crystal structure from the Protein Data Bank. Aptamer sequences that could bind to the SARS-CoV-2 RBD were obtained from a previous report.
Ultimately, the CoV2-RBD-1 (or RBD-1C) aptamer was selected as the parent sequence for the generation of mutated sequences. The mutated sequences were then generated, whereas the ZDOCK docking program was used to dock the S protein with the two three-dimensional (3D) aptamer models.
A comparison of the secondary structure of the mutated sequence with the RBD-1C aptamer was carried out. The ZRANK scoring function was also used along with ZDOCK to determine interactions between the aptamer and target protein molecule. Finally, three-dimensional (3D) models of the aptamers were obtained for molecular docking (MD) simulations.
Study findings
The ZDOCK and ZRANK scores for the best docking pose of the RBD-1C aptamer were 44.4 and −88.392. Of the 223 amino acids in the RBD of the S1 subunit, 24 were identified to be present on the binding interface between the S protein and the aptamer. Moreover, 18 of the 595 mutated sequences were found to have lower ZRANK scores as compared to the threshold value.
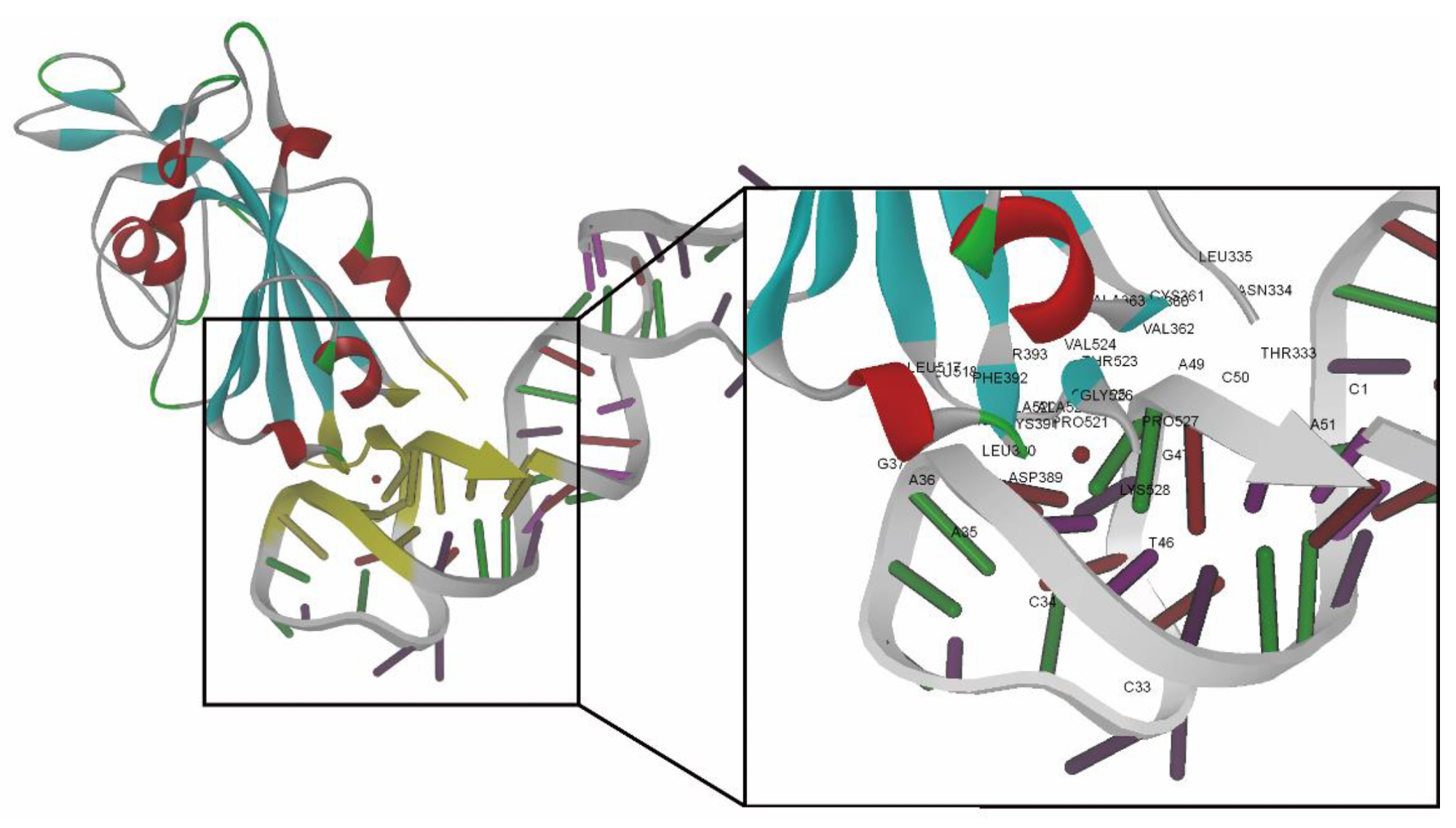 Best docking pose of RBD-1C aptamer against the S protein (visualization software: DS 4.1). Amino acids and nucleotides involved in the binding interface of the S protein/RBD-1C complex are marked with yellow. The close-up view presents the amino acids and nucleotides in the binding interface marked by labels.
Best docking pose of RBD-1C aptamer against the S protein (visualization software: DS 4.1). Amino acids and nucleotides involved in the binding interface of the S protein/RBD-1C complex are marked with yellow. The close-up view presents the amino acids and nucleotides in the binding interface marked by labels.
The two best-selected aptamers for the S protein were named RBD-1CM1 and RBD-1CM2. The RBD-1CM1 aptamer was reported to have two mutated nucleotides at positions 34 and 47, while the RBD-1CM2 aptamer had two mutated nucleotides at positions 41 and 44.
The root-mean-square deviation (RMSD) obtained in the MD simulation suggested that the RBD-1CM1 aptamer could be a better recognition element for the S protein as compared to the other two aptamers.
The total energies of the aptamer and S protein complexes were negative for all three aptamers. This suggests that the complexes were stable systems, where the S protein/RBD-1CM2 complex had the highest stability, followed by the S protein/RBD-1C complex and S protein/RBD-1CM1 complex.
The average number of hydrogen bonds was 2.89, 5.39, and 3.15 for the S protein/RBD-1C, S protein/RBD-1CM1, and S protein/RBD-1CM2 complexes, respectively. Each complex had a maximum number of ten, 14, and ten hydrogen bonds, respectively.
The most stable complex concerning hydrogen bonds was suggested to be the S protein/RBD-1CM1 complex. Additionally, the binding affinity values for RBC-1CM1 and RBC-1CM2 aptamers were equal and slightly more than that of the RBD-1C aptamer.
Taken together, the current study determined that the RBD-1CM1 aptamer is the best aptamer for the SARS-CoV-2 S protein. This aptamer can be used as an alternative to antibodies and have several applications in the diagnosis and treatment of SARS-CoV-2.
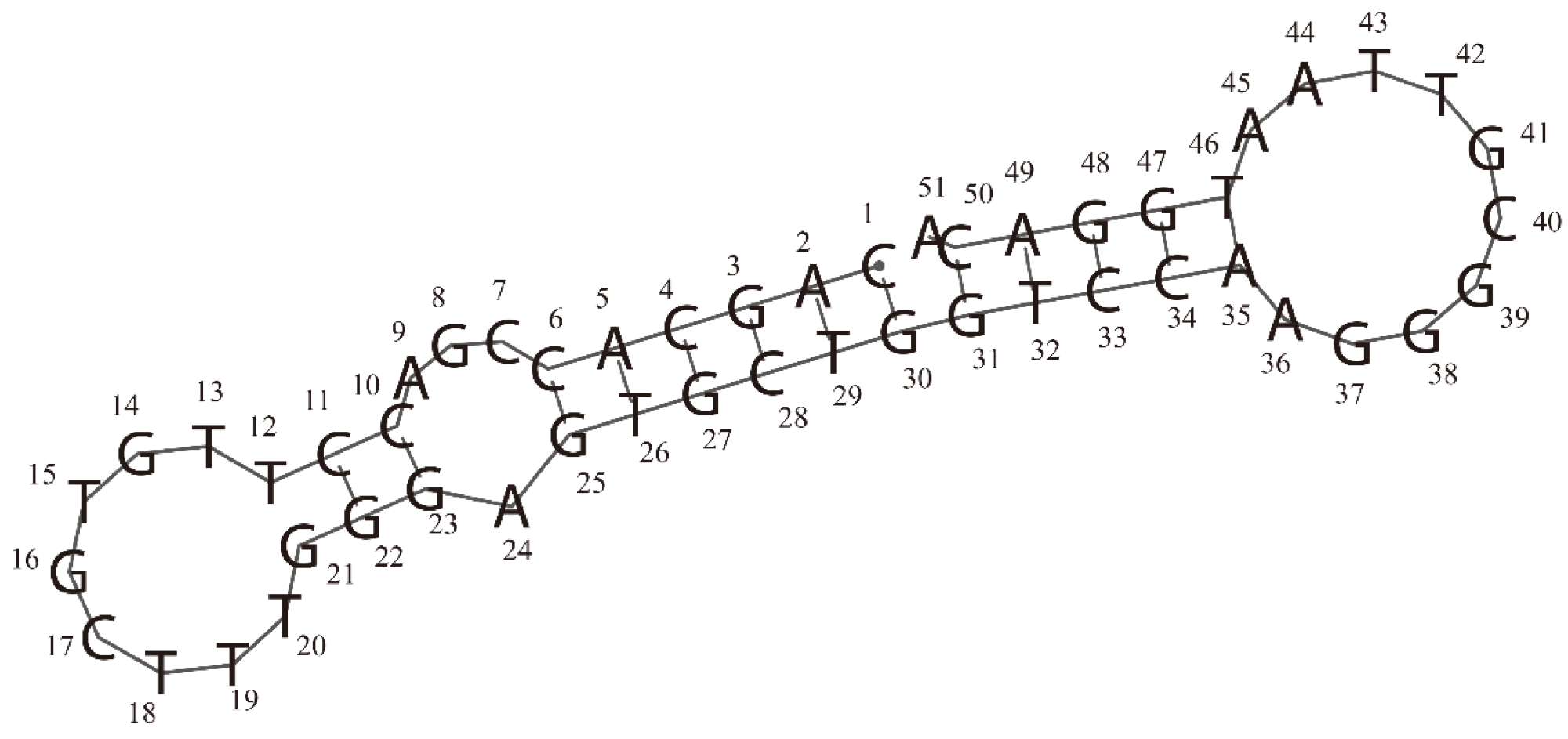 Secondary structure of the RBD-1C aptamer was predicted by the RNAfold web server. In this structure, 2 hairpins (at two ends) and 1 internal loop can be observed.
Secondary structure of the RBD-1C aptamer was predicted by the RNAfold web server. In this structure, 2 hairpins (at two ends) and 1 internal loop can be observed.
Limitations
The study had two limitations. First, only the S protein RBD is adopted in the study. Second, computing power limits the simulation time of MD.
Journal reference:
- Lin, Y., Chen, W., Hwu, E., & Hu, W. (2022). In-Silico Selection of Aptamer Targeting SARS-CoV-2 Spike Protein. International Journal of Molecular Sciences 23(10). doi:10.3390/ijms23105810.
 Large Swedish study finds COVID-19 vaccination unrelated to fertility or childbirth rates
Large Swedish study finds COVID-19 vaccination unrelated to fertility or childbirth rates