In a recent study posted to the bioRxiv* pre-print server, researchers studied the sequence data from the coronavirus disease 2019 (COVID-19) cases from the Huanan seafood market in Beijing, China, to define the minor variant genome populations from the start of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-induced pandemic.
 Study: SARS-CoV-2 minor variant genomes at the start of the pandemic contained markers of VoCs. Image Credit: NIAID
Study: SARS-CoV-2 minor variant genomes at the start of the pandemic contained markers of VoCs. Image Credit: NIAID
 *Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Background
The Huanan market in China was the epicenter of the COVID-19-induced pandemic, where human-to-human transmission of the lethal SARS-CoV-2 began. Two spillover events at the market established transmission of lineages B and A in the human population and potentially ended other spillovers. Consequently, the researchers have believed that SARS-CoV-2 had limited genetic diversity at the start of the pandemic in humans. However, as the pandemic progressed and SARS-CoV-2 disseminated within China and globally, its genomic sequences became more and more diverse.
The dominant genome sequence is the most abundant in a sample from a COVID-19 patient and a useful marker of sequencing information of viral populations. For example, in the patients from the Huanan market, the dominant SARS-CoV-2 genome sequences were over 99.9% identical, indicating the recent emergence of SARS-CoV-2 into the human population. The second markers are the minor genomic variants with a lower abundance. They contain synonymous and non-synonymous amino acid substitutions that confer an advantage under selective pressure and thus affect the disease phenotype.
About the study
In the present study, researchers identified the earliest samples of COVID-19 patients reportedly associated with the Huanan market based on the symptom onset or sequence deposition date. They identified SARS-CoV-2 sequencing data from 16 patients (S1 to S16) and used different approaches to assign minor genome variants from this data.
The researchers separated the low-frequency variants from Illumina sequence errors using DiversiTools, whose algorithms use the Illumina quality scores to calculate a p-value for each variant at each nucleotide site in a protein. Alternatively, they considered an arbitrary read depth of 10 or 100 nucleotides per site to identify minor variant genomes. The researchers considered a cut-off greater and lower than the 20% threshold to investigate minor genomic variants and their association with amino acid substitutions and phenotype.
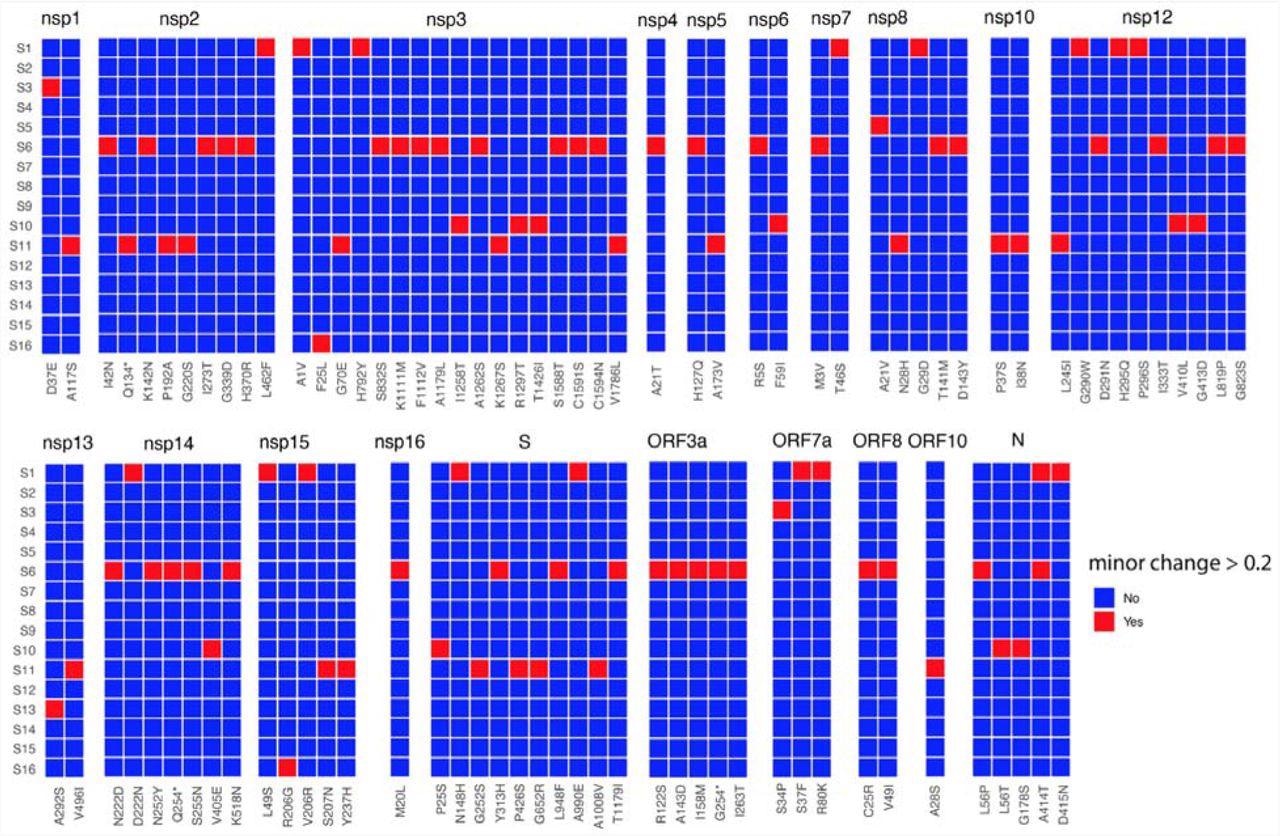
Heat map of non-synonymous changes at the minor variant level in SARS-CoV-2 that have a threshold between 20 and 49% in the 16 patients (y-axis). The panel is divided into each of the SARS-CoV-2 proteins and substitutions are shown on the x-axis. The amino acid site with coverage >= 10 are shown.
Study findings
The authors noted that the coverage of the minor variants was variable across the genome, with some sites having higher and lower coverage in the sequencing data. Further, they identified minor variant genomes across all the genes in SARS-CoV-2 for each patient. The data indicated that amino acid substitutions at specific sites in some genes, including the SARS-CoV-2 envelope (E), membrane (M), open reading frame (ORF)6, ORF7b, and ORF10, were less frequent.
Notably, three patients, S9, S12, and S14, had little population diversity in minor variant genomes in SARS-CoV-2. Conversely, patient S6 possessed ~40% (higher) frequency of substitutions, C25R and V49I in the SARS-CoV-2 ORF8 gene. For a threshold of over 20%, patients S1, S6, S10, and S11 had a greater number of minor variant genomes in SARS-CoV-2. Moreover, these genomes had premature stop codons (e.g., patient S11 had a premature stop codon in SARS-CoV-2 non-structural protein 2 (NSP2).
However, more importantly, the authors identified several minor genomic variants in the SARS-CoV-2 spike (S) and other proteins, subsequently found across its variants of concern (VOCs). The frequency and position of these genomic variants varied across patients. Nevertheless, the data indicated the presence of these hallmarks of SARS-CoV-2 VOCs at the start of the pandemic. For instance, the P323L substitution in NSP12 in SARS-CoV-2 minor genomic variants from patients S3, S9, S10, and S15.
Moreover, although these substitutions were under 5% at a minor variant genome level, they were found to be under strong selection pressure. Accordingly, they had the potential to grow in dominance within days of SARS-CoV-2 infection. Notably, minor genomic variants of SARS-CoV-2 from patient 16 had several substitutions in a frequency range of 5 and 15%.
Conclusions
The current study identified amino acid substitutions within the minor variants in SARS-CoV-2 cases at the start of the COVID-19 pandemic that was associated with future VOCs. Furthermore, it showed the possibility of predicting the emergence of VOCs based on the analysis of minor variant genomes and the variable sites from the start of the COVID-19 outbreak. To conclude, these insights into the viral evolution could help devise treatment regimens and vaccination against future SARS-CoV-2 VOCs on time.
*Important notice: bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
 Large Swedish study finds COVID-19 vaccination unrelated to fertility or childbirth rates
Large Swedish study finds COVID-19 vaccination unrelated to fertility or childbirth rates