Background
SARS-CoV-2, the pathogen responsible for COVID-19, has accumulated multiple mutations since it was first identified in December 2019. These mutations have resulted in more transmissible and resistant SARS-CoV-2 variants, several of which have been categorized by the World Health Organization (WHO) as variants of concern (VOCs).
The SARS-CoV-2 Omicron variant was first detected in South Africa on November 9, 2021, and was subsequently declared a VOC by the WHO on November 26, 2021. The Omicron spike protein consists of 26-32 mutations compared to previously sequenced viral isolates.
Since its initial detection, Omicron has evolved into five distinct sub-lineages, including BA.1, BA.2, BA.3, BA.4, and BA.5. Many of these Omicron subvariants have been responsible for the recent waves of reinfections and vaccine breakthrough infections.
Viral mutations can be categorized as synonymous or non-synonymous. Whereas synonymous mutations do not impact the protein sequence, non-synonymous cause changes in the viral proteome. Despite the apparent neutrality of synonymous mutations, their ability to accumulate over time can lead to altered protein expression profiles.
Widespread SARS-CoV-2 mutations
The 97,437 SARS-CoV-2 protein sequences analyzed in the current study identified non-synonymous mutations in 26 different sequences. Therefore, the researchers were particularly interested in assessing whether mutations acquired in replication-associated SARS-CoV-2 proteins affected viral replication processes. These included non-specific protein 7 (Nsp7), Nsp8, Nsp12, Nsp13, and Nsp14, which were associated with replication of the Omicron variant, and were analyzed for acquired mutations.
The P314L mutation in Nsp12, a ribonucleic acid (RNA)-dependent RNA polymerase, was identified in almost 98% of the analyzed SARS-CoV-2 protein sequences. However, this mutation was not unique among the Omicron sub-lineages; thus, it does not appear responsible for the increased number of mutations in these new viral variants.
Other commonly identified mutations included R392C in Nsp13, which is an RNA-helicase, as well as I42V in Nsp14, which is a 3’-5’ exoribonuclease protein. However, both mutations were not located near the active site of either protein, thus indicating that they similarly did not significantly contribute to the evolution of Omicron subvariants.
Further analysis demonstrated that the SARS-CoV-2 structural spike, envelope, membrane, and nucleocapsid proteins, as well as the accessory proteins, including open reading frame 7a (ORF7a), ORF7b, ORF9, and ORF9b, have evolved more rapidly as compared to the non-structural proteins.
How did Omicron evolve?
Although the mechanisms by which Omicron emerged remain unclear, it is suspected that this variant evolved in immunocompromised patients. The evolution of SARS-CoV-2 within these patients could be due to their weak immune systems that were unable to clear the virus but strong enough to allow for variants of increased fitness to proliferate.
The emergence of the Omicron variant may have occurred due to a strong purifying selection on NSP1-16 and a strong diversifying selection on the SARS-CoV-2 structural and accessory proteins. Another possible reason could be that Omicron represents a recombinant version of the SARS-CoV-2 ancestral strain and a hyper-mutated strain that has yet to be identified.
Nevertheless, further research is needed to elucidate whether each of these scenarios is, in fact, responsible for the emergence of the Omicron variant.
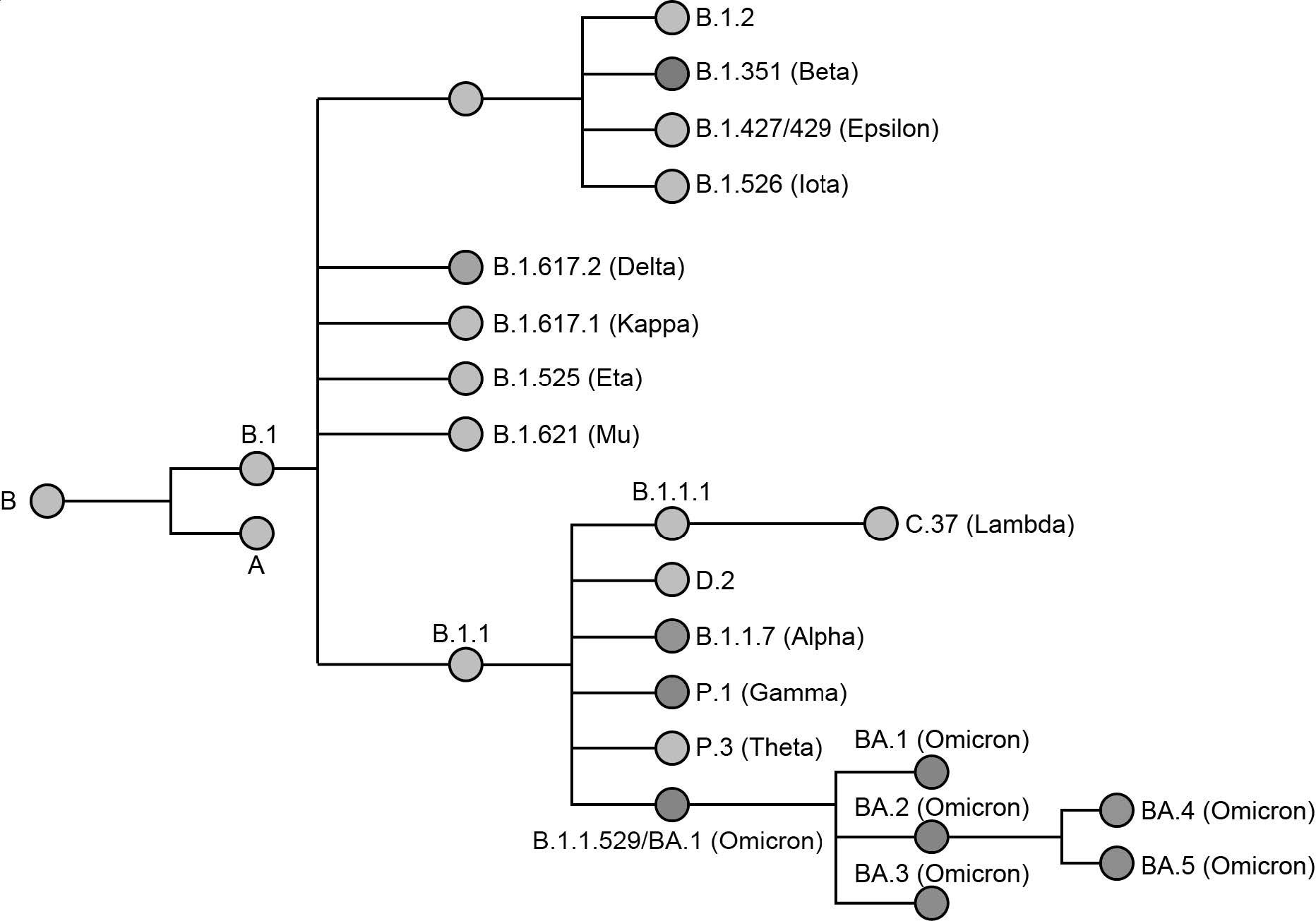 Phylogenetic relationship of named SARS-CoV-2 variants. Variants of concern (VOC) are represented by a colored node. The phylogenetic tree was adapted from data provided by NextStrain, CoVariants (i.e., covariants.org), and Pangolin (i.e., cov-lineages.org)
Phylogenetic relationship of named SARS-CoV-2 variants. Variants of concern (VOC) are represented by a colored node. The phylogenetic tree was adapted from data provided by NextStrain, CoVariants (i.e., covariants.org), and Pangolin (i.e., cov-lineages.org)
Future outlook
Currently, the diagnosis of COVID-19 is primarily based on detecting the presence of the nucleocapsid protein or gene through antigen and polymerase chain reaction (PCR) assays, respectively. If the nucleocapsid protein continues to accumulate mutations, the diagnosis of COVID-19 in the future could be compromised.
Mutations in the spike protein, which remains the primary target of current COVID-19 vaccines and therapeutics, will also significantly affect vaccine-induced and natural immunity. Thus, new vaccines that consist of the spike protein of different SARS-CoV-2 variants could improve the durability of immune responses following immunization.
Despite the widespread distribution of vaccines, COVID-19 cases continue to rise, particularly as new Omicron subvariants have been identified. Thus, adequate viral surveillance through patient diagnoses and wastewater sampling is imperative to mitigate the threat of new SARS-CoV-2 variants.
The authors conclude, "There is no easy solution to this situation, but as infections increase, and tracking decreases, we are wandering blind into an uncertain future and we are at greater risk of being caught off guard by an unlucky role of the evolutionary dice, which may result in a new variant with distinct clinical outcomes."
Journal reference:
- Wiegand, T., Nemudryi, A., Nemudraia, A., et al. (2022). The Rise and Fall of SARS-CoV-2 Variants and Ongoing Diversification of Omicron. Viruses 14(9);2009. doi:10.3390/v14092009.
 CDC tracks SARS-CoV-2 BA.3.2 global rise and finds early signals in U.S. wastewater
CDC tracks SARS-CoV-2 BA.3.2 global rise and finds early signals in U.S. wastewater