Two types of rare genetic diseases of innate immunity have been identified in humans. One is rare inborn errors of type I interferon (IFN) immunity due to the variants of genes governing type I IFN immunity. The other disease is rare inborn errors of development of autoantibodies against type I IFNs due to variants of the autoimmune regulator gene (AIRE) controlling T cell tolerance.
The enormous amount of research conducted during the COVID-19 pandemic has discovered that these two inborn errors of type 1 IFN immunity account for approximately 15-20% of severe COVID-19-related pneumonia cases in unvaccinated individuals.
Inborn errors of type 1 IFN immunity and viral infections
Humans have so far been identified as having 21 types of inborn errors of type I IFN immunity. The first discovery was made in 2003 in a child with herpes simplex virus encephalitis who represented autosomal recessive (AR) complete STAT1 deficiency. This deficiency is associated with the loss of type I, II, and III IFN responses, predisposing patients to many viral diseases.
The incidence of AR complete STAT2 deficiency and AR complete IFN regulatory factor 9 deficiency is relatively low. Nevertheless, these conditions predispose patients to a narrow range of viral infections.
Inborn errors of IFN-α/β receptor chains 1 and 2 (IFNAR1 and IFNAR2) deficiencies have also been identified. These patients are susceptible to only a few viral infections, with herpes simplex virus encephalitis and acute influenza being the most commonly detected infections. These deficiencies make individuals resistant to most common viral infections. These observations indicate that type I IFN immunity is required to eliminate only a small range of viruses and that there are type I IFN-independent mechanisms to induce broad-spectrum antiviral immunity.
Inborn errors of JAK1 and TYK2, constitutively associated with IFNAR1 and IFNAR2, respectively, have been identified. Patients with AR JAK1 deficiency are susceptible to a few viral infections due to impaired type I IFN immunity. In a certain fraction of patients with AR TYK2 deficiency, impaired responses to interleukin 23 (IL-23) have been noticed, which predisposes them to mycobacterial infections. Type I IFN response is partially affected in these patients, and type III IFN response is mainly maintained.
The induction of type I IFN responses to viral infection depends on a range of transcription factors and regulators, including NEMO, TBK1, IRF3, and IRF7. AR deficiencies of these proteins have been identified. Patients with AR IRF7 deficiency are susceptible to developing severe influenza pneumonia. In contrast, patients with autosomal dominant (AD) IRF3 or TBK1 deficiency are susceptible to herpes simplex virus encephalitis.
Other inborn errors of type 1 IFN immunity include AR deficiencies of TLR7, IRAK4, and MyD88. Patients with these deficiencies are at higher risk of critical COVID-19 pneumonia.
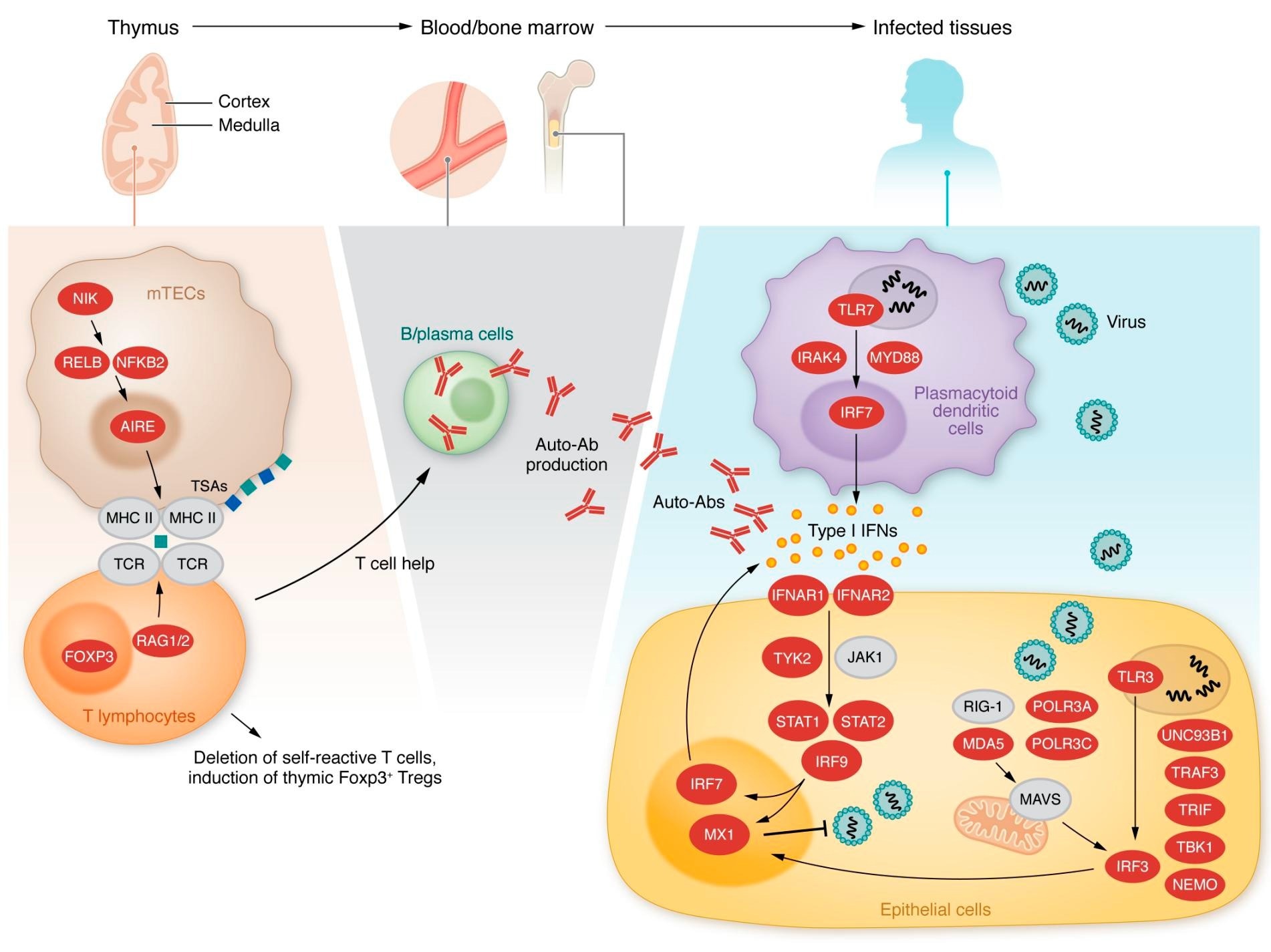 Inborn errors of type I IFN immunity or tolerance. Left, middle: Variants in genes expressed in thymic medullary epithelial cells, indicated in red, are linked to a defect in T cell selection and the production of type I IFN autoantibodies. Right: Variants in genes indicated in red alter type I IFN induction and response pathways.
Inborn errors of type I IFN immunity or tolerance. Left, middle: Variants in genes expressed in thymic medullary epithelial cells, indicated in red, are linked to a defect in T cell selection and the production of type I IFN autoantibodies. Right: Variants in genes indicated in red alter type I IFN induction and response pathways.
Inborn errors of type 1 IFN tolerance
Besides inborn errors of type I IFN-related genes, autoantibodies targeting type I IFNs have also been discovered. These autoantibodies impair the activity of IFNs. The condition is medically termed autoimmune polyglandular syndrome type 1 (APS-1) and is characterized by the development of multiple organ-specific autoimmune diseases.
Inborn defects in the AIRE gene are the main etiology of this condition. In addition, studies have shown that AIRE promotes the expression of multiple tissue-specific self-antigens and controls T-cell immune tolerance.
Type I IFN deficiency and severe COVID-19
The COVID Human Genetic Effort was initiated during the pandemic to identify a causal link between single-gene inborn errors of immunity and the development of critical COVID-19 pneumonia, which is responsible for the majority of COVID-related deaths.
The initial hypothesis made by the scientific community was that critical pneumonia due to the seasonal influenza virus and critical pneumonia due to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) might be allelic.
Three core influenza susceptibility genes, including IFR7, IRF9, and TLR3, have been selected for the analysis. In addition, another ten genes related to these core influenza genes and germline variants related to other critical viral infections have been included.
The findings of the genetic analysis revealed that tonic type I IFN levels in respiratory epithelial cells (RECs) is vital for host defense against SARS-CoV-2. A causal link has been identified between AR deficiencies in IFR7, IRF9, and TLR3 and COVID-19 pathogenesis.
Furthermore, X-linked recessive TLR7 deficiency has been observed in 1-2% of male patients with critical COVID-19 pneumonia. Recessive deficiency in TLR7 has also been observed in 10% of children with COVID-19 pneumonia. Mechanistically, rare variants of TLR7 have been found to impair the recruitment of dendritic cells to the respiratory tract in response to SARS-CoV-2 infection, leading to the loss of type I IFN-mediated protective immunity.
The presence of pre-existing autoantibodies against type I IFN has been implicated in critical COVID-19 pathogenesis. Critical COVID-19 pneumonia has been observed in several patients with APS-1. These autoimmune antibodies have been found to delay type I IFN-stimulated gene response in leukocytes and nasal mucosa. The analysis of pre-pandemic blood samples revealed that the prevalence of these antibodies remains stable until the age of 65 years and subsequently increases after the age of 80.
Screening of autoantibodies against type I IFNs would, thus, be vital for determining the prognosis of COVID-19. Studies have found that in 20% of vaccinated individuals, hypoxemic pneumonia occurs due to these autoantibodies.
Recent evidence has indicated that type I IFN-targeting autoantibodies are responsible for herpes simplex virus infection in hospitalized COVID-19 patients. Overall, a significant involvement of IFN-targeting autoantibodies has been established in four viral diseases, including critical COVID-19 pneumonia, influenza pneumonia, adverse reactions to the yellow fever virus (YFV-17D) vaccine, and recurrent or disseminated shingles caused by the varicella-zoster virus.
 Why some people can’t lose weight even when they follow the rules
Why some people can’t lose weight even when they follow the rules