The study data could aid diagnosis and drug resistance (DR) mapping of tuberculosis (TB) patients for infection control.
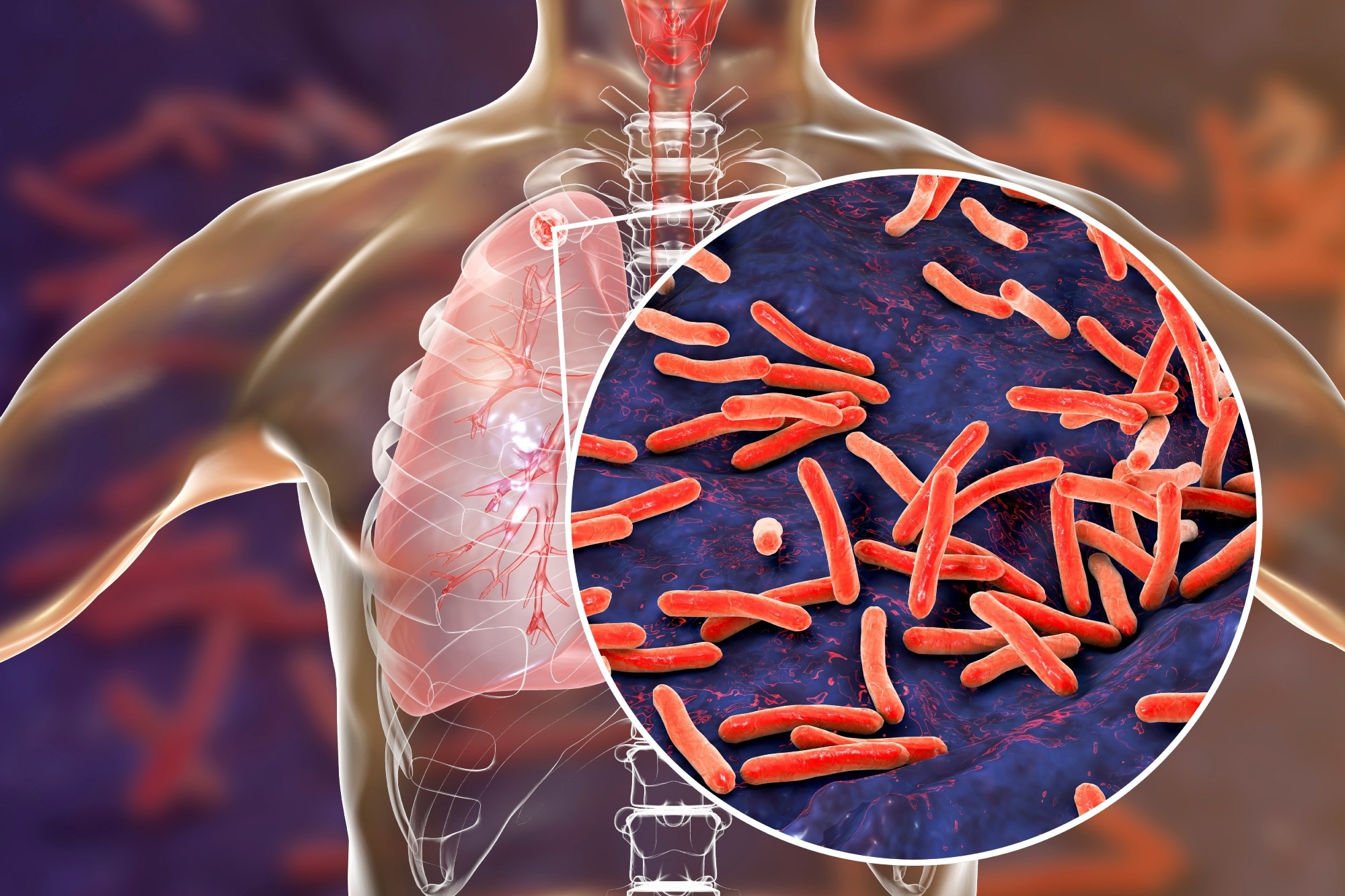 Study: Mixed infections in genotypic drug-resistant Mycobacterium tuberculosis. Image Credit: Kateryna Kon/Shutterstock.com
Study: Mixed infections in genotypic drug-resistant Mycobacterium tuberculosis. Image Credit: Kateryna Kon/Shutterstock.com
Background
M. tuberculosis, the causal agent of TB, has four lineages (L1-L4), each having several strains with varying transmissibility and disease-causing potential.
Clinical studies have reported that some TB patients harbor multiple M. tuberculosis strains, resulting in within-host MSIs.
The presence of drug-susceptible and drug-resistant strains within the same host contributes to multi-drug resistance (MDR-TB), which hinders infection control by first-line TB treatment, rifampicin (RR-TB) and isoniazid (HR-TB) and also contribute to the spread of drug-resistant strains.
Yet studies have barely identified drug-resistant strains within MSI samples of M. tuberculosis.
About the study
The present study analyzed 50,723 M. tuberculosis isolates where WGS and drug susceptibility test (DST) data were publicly available.
These samples, collected from 64 countries, exhibited ≥99% genome-wide coverage and sequencing read depths of 30-fold or higher. Moreover, these samples encompassed all major M. tuberculosis lineages, with respective proportions of L1, L2, L3, and L4 being 9.1%, 27.6%, 11.8%, and 48.3%.
The TB-Profiler software initially detected MSIs and inferred genotypic drug resistance in these samples, including the supported read coverage of sub-lineages within each sample.
Notably, it used different informative mutation lists for genotypic profiling. Next, the researchers built a GMM for each sample and assessed its performance. GMM helped detect mixed gene reads and MSIs across all M. tuberculosis sub-lineages.
Performance measures included the mean square error (MSE) and the accuracy of the drug resistance profiling compared to TB-Profiler predictions.
In addition, they used WGS data from 48 samples from clinical Malawi M. tuberculosis strains to evaluate GSS performance on artificial mixes of bacterial deoxyribonucleic acid (DNA) samples.
Results
The results showed that M. tuberculosis lineage 2.2.1 was the most prevalent in four, Southeast Asia, the Western Pacific, Africa, and Europe of six World Health Organization (WHO) regions evaluated.
TB-Profiler software predicted a significant proportion of M. tuberculosis isolates were resistant to isoniazid and rifampicin; accordingly, these were MDR-TB. In addition, genotypic resistance predictions were consistent in most cases.
Further, the researchers noted that the L4 strain and L4.3.3 sublineage were the most prevalent globally. Genotypic drug resistance was the highest in the Eastern Mediterranean region, predominantly due to L3 strains.
M. tuberculosis MSIs are informative for heteroresistance, which can mitigate the effectiveness of TB treatment. TB-Profiler software predicted MSIs in 531/48,679 samples, i.e., in 1% samples. Quant-TB software confirmed most MSIs identified by TB-Profiler.
While Lineage 4 M. tuberculosis strains were the most involved in MSIs, La1.1, L2.2, and M. caprae showed some involvement, too. In combination, L4 and L2 caused MSIs, reflecting the confounding effects of the sampling.
Furthermore, the GMM approach revealed decreased involvement of the less transmissive lineages, e.g., M. tuberculosis lineage 7, likely because their sequencing rates are relatively low.
GMM models and TB-Profiler attained low MSEs in samples with a dominant strain, indicating good predictive power.
Their performance was good since the overall MSE values were consistently low for both methods. Notably, Quant-TB attained a higher overall MSE value than the other methods.
Conclusions
M. tuberculosis culture & colony sampling techniques and bioinformatic analyses (used earlier) underestimated the degree of MSIs in M. tuberculosis samples.
On the contrary, direct WGS of sputum or lung tissue presented a better and more accurate representation of M. tuberculosis diversity within a TB patient. These methods have also shown that TB infections are way more complex than previously thought.
In the current study, combining WGS data with the GMM approach, a non-culture-based method for drug resistance profiling effectively predicted the relative abundance of different strains in DNA samples with known DR and mixing proportions.
GMM models predicted highly accurate drug resistance across the minor mixing proportions, i.e., those between 0.05 and 0.50, with an overall MSE of 0.012. On the contrary, MSE values for TB-Profiler and Quant-TB were a little lower (0.009) and greater (0.013).
Overall, GMM data could be insightful for clinical decision-making in TB cases, aid diagnosis, and optimize the personalization of treatments. Most importantly, WGS-based diagnoses for TB could help avoid ineffective drug(s) use.
Further improvements to the GMM approach through the use of M. tuberculosis phylogenetic tree structure could even extend the benefits of this approach to other MTBC members, such as M. bovis, and M.caprae, to name a few.
 Unmasking the impact of C. neoformans as a Global Menace
Unmasking the impact of C. neoformans as a Global Menace