The severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) genome harbors a single-stranded, positive-strand ribonucleic acid genome (ssRNA (+) genome). This ssRNA (+) genome is between 26-32 kilobases (kb) in size, with high fidelity replication due to a 3′-to-5′ exoribonuclease 'proof-reading' mechanism.
Since its original emergence in December 2019, the SARS-CoV-2 genome has continued to evolve and exhibit quasi-species diversity as it adapts to its human host. While most of the thousands of mutations identified in the SARS-CoV-2 genome, to date, are not significant functional changes, they have proven etiologically valuable.
Study: Cooccurrence of N501Y, P681R and other key mutations in SARS-CoV-2 Spike. Image Credit: Design_Cells / Shutterstock.com
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Background
To date, the SARS-CoV-2 has mutated into five variants of concern (VOCs). The essential spike mutation 'N501Y' is shared by Alpha, Beta, Gamma, and Omicron VOCs, whereas the 'P681R' is critical to Delta's dissemination.
Since the onset of the coronavirus disease 2019 (COVID-19) pandemic, researchers had predicted that significant mutations would occur, particularly in the spike protein. The D614G mutation announced in April 2020 resulted in the 'G15 strain' with higher infectivity replacing the genetic background of this virus globally, while there was no impact on vaccine efficacy.
Nevertheless, the N501Y mutation shared by the Alpha, Beta, Gamma, and Omicron variants VOCs has rendered increased infection rates and transmission, decreased vaccine efficacy, as well as enhanced the ability of SARS-CoV-2 to infect novel species, such as wild-type mice. Another significant mutation is P681R, which changes the furin cleavage site and is responsible for the Delta variant's greater infectivity, transmission, and worldwide impact.
About the study
Considering these findings, a recent study published on the medRxiv* preprint server evaluates the risk of the P681R, N501Y, and D614G mutations of current interest (MOCI) co-occurring naturally due to convergent evolution. This has led to SARS-CoV-2 variants that are of greater concern as compared to those identified previously, noting that the recent Omicron VOC has the N501Y but not the P681R mutation.
The current study also aims to establish a methodology and pipeline for analyzing the propagation of variants harboring significant mutations in combination.
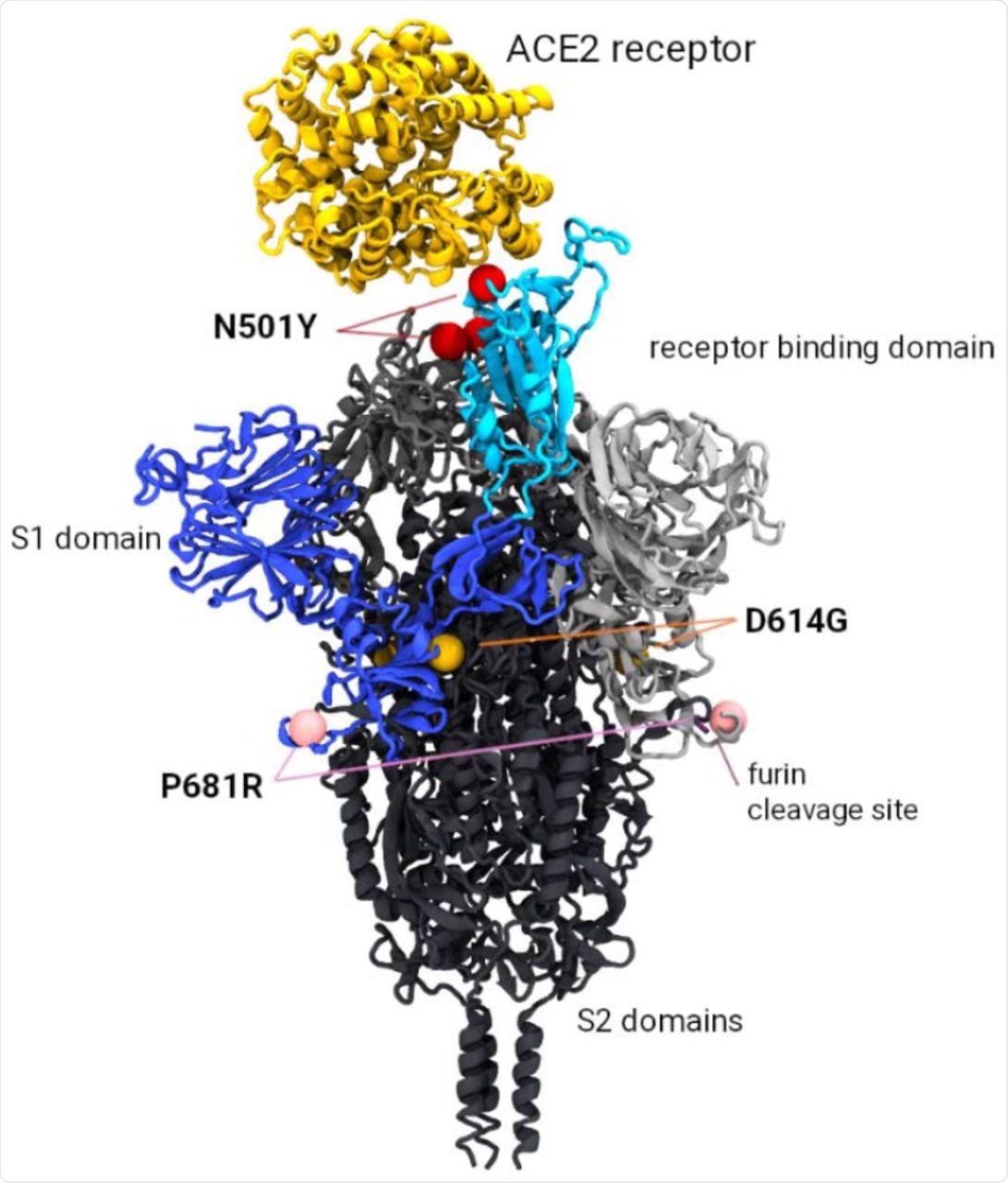
Model of the SARS-CoV-2 Spike trimer protein indicating the relative position of the P681R, N501Y and D614G mutations of current interest (MOCI).
Herein, data was extracted from the Global Initiative for the Sharing of All Influenza Data (GISAID), the world's largest and most complete archive of SARS-CoV-2 sequences. The study employed quasi-species theory and in-silico modeling to evaluate findings.
The paper analyzed over 4.2 million SARS-CoV-2 genomic sequences from the GISAID and established that N501Y and P681R mutations co-occurred at least 3,678 times on the spike D614G mutation background between October 2020 and November 2021. The first observation of these three MOCI was made in Slovenia on October 17, 2020; however, no subsequent occurrences were reported in that country.
Study findings
GISAID data mining revealed 3,678 entries (0.1%) contained the MOCI, the majority of which were in the Alpha (61.3%) variants, as well as the Beta (0.4%), Gamma (6.4%), Delta (21.2%), and Omicron (0.0%) strains. Furthermore, a minority of these MOCI were recognized in the current or former variants of interest (VOI), including the Mu (0.3%), Kappa (0.2%), and Iota (0.02%) strains. A small proportion of 10.2% was also detected in other variants not classified as VOC, VOI, Variant Under Monitoring (VUM), or specific sub-lineages.
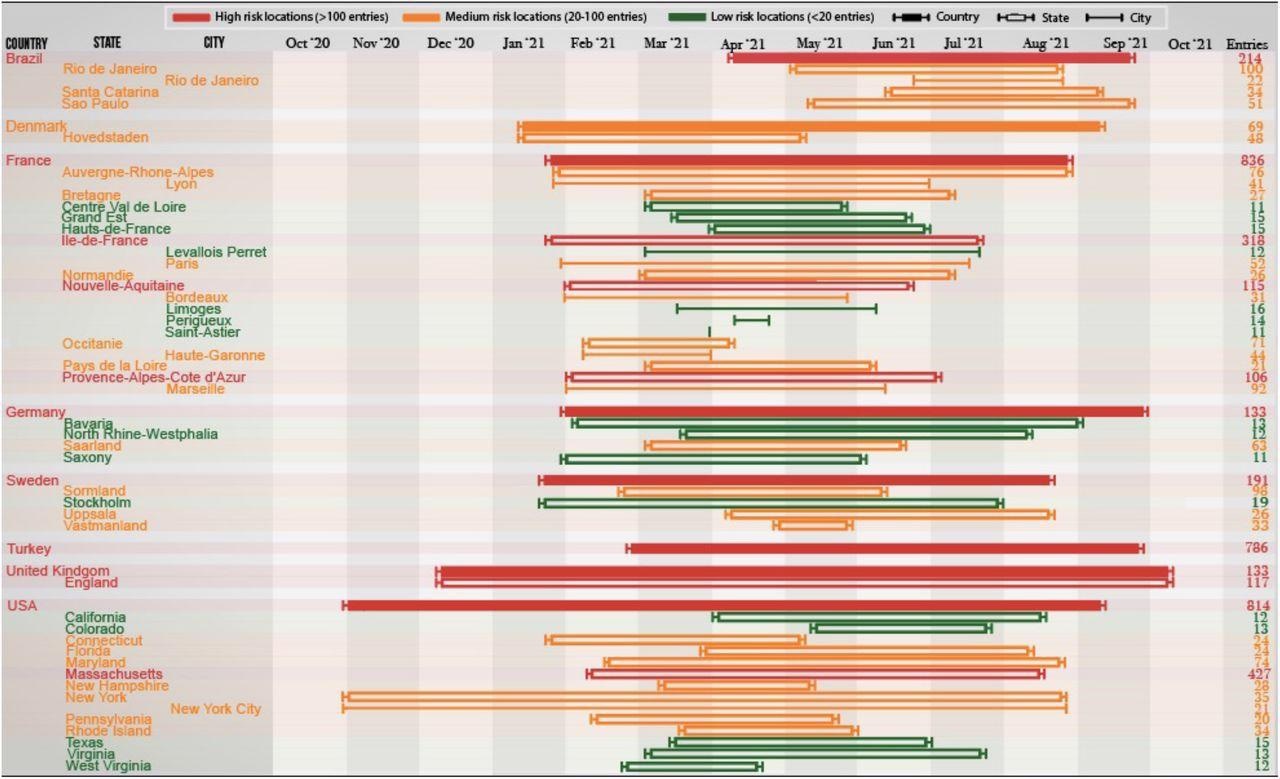
Discrete timeline depicting the start and end which SARS-CoV-2 variants with the P681R, N501Y and D614G mutations were observed in various countries, states and cities between October 2020 and October 2021. Countries with 50 or more observations, and states/cities with 10 or more observations are deemed significant and thus included in this timeline.
The Y501-H681 combination was found in around 1.1 million entries in both the Alpha and Omicron VOCs. Two-thirds of the 3,678 co-occurrences were recorded in France, Turkey, or the United States of America, while others were reported in 57 other countries.
Moreover, 55.5% and 39.6% of co-occurrences involved Alpha Q.4 and Gamma P.1.8 sub-lineages, respectively, acquired the P681R. Meanwhile, 10.7% and 3.8% involved Delta B.1.617.2 lineage and AY.33 sub-lineage, respectively, acquired the N501Y, whereas the remaining 10.2% involved other variants. N501Y and P681R individually rendered selective advantages; however, the Y501-R681 combination did not outcompete other variations in all tested instances.
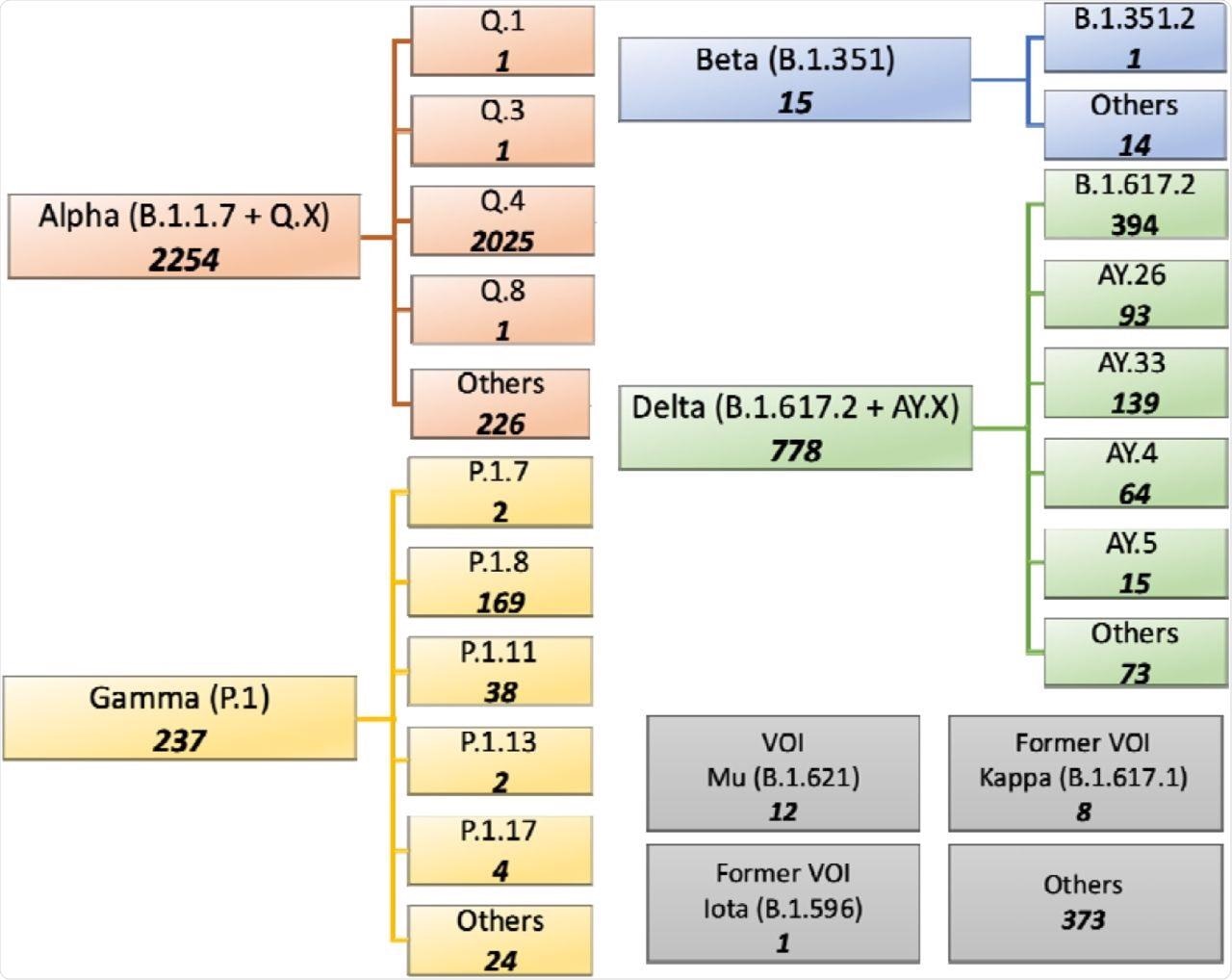
Summary of SARS-CoV-2 lineages and sub-lineages containing the D614G, N501Y and P681R mutations of current interest (MOCI) on GISAID.
Implications
While the current study's findings are encouraging for global public health initiatives, in vitro, and in vivo studies are required in the absence of compelling in silico explanations for this event. The current study also established a pipeline for systematically analyzing combinations of important mutations from public domain data and providing early warnings of spread.
Although predicting a specific combination of mutations that would provide the best selective advantage remains a challenge, mutations previously observed in other dominant lineages may reemerge spontaneously in contemporary strains, thus increasing their infectivity and worsening disease prognosis.
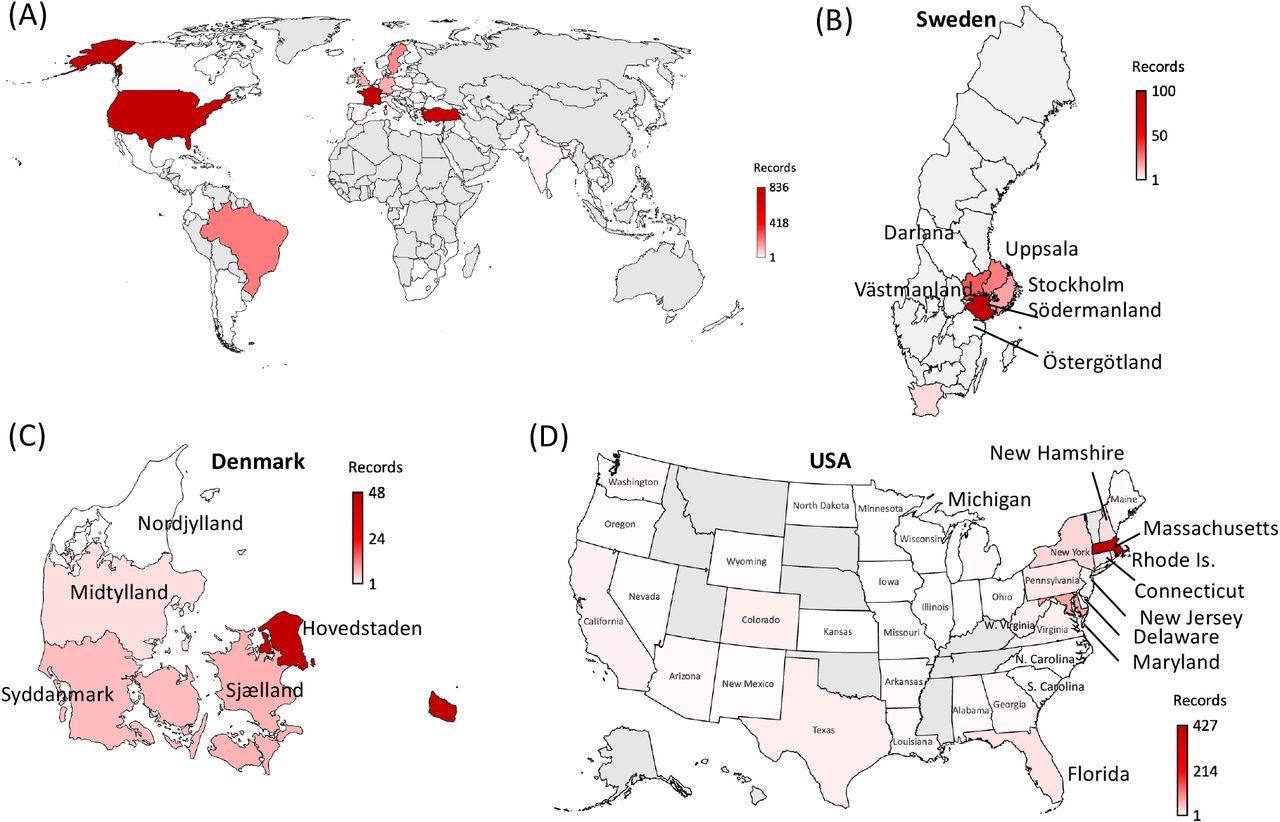
World map depicting the 65 countries where SARS-CoV-2 variants containing the cooccurring MOCI were recorded between October 2020 and October 2021. (A) world map, with zoomed-in view of the regions observed with MOCI shown for (B) Sweden (C) Denmark and (D) USA. Countries are color-coded based on the number of records of MOCI observed, as shown in the legend, with greyed-out regions indicating no observations. For some countries such as Turkey, region/city-level information was not available, but EP _ISL numbers can be provided upon request.
The present study found that the co-occurring three key mutations of D614G, N501Y, and P681R do not appear to spread. Despite being identified in three countries, the Y501-R681 combination has not outcompeted other variants.
The methodology used in the current study could have potential use in determining the transmissibility of certain combinations of mutations, specifically in the Omicron variant, which harbors multiple un-identified mutations.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Lee, C., Mangalaganesh, S., Wilson, L. O. W., et al. (2021). Cooccurrence of N501Y, P681R and other key mutations in SARS-CoV-2 Spike. medRxiv. doi:10.1101/2021.12.25.21268404. https://www.medrxiv.org/content/10.1101/2021.12.25.21268404v1.
- Peer reviewed and published scientific report.
Lee, Carol, Shruthi Mangalaganesh, Laurence O. W. Wilson, Michael J. Kuiper, Trevor W. Drew, and Seshadri S. Vasan. 2022. “Tracking Co-Occurrence of N501Y, P681R, and Other Key Mutations in SARS-CoV-2 Spike for Surveillance.” Zoonotic Diseases 2 (3): 147–62. https://doi.org/10.3390/zoonoticdis2030014. https://www.mdpi.com/2813-0227/2/3/14.
 Long COVID symptoms change for 15 months after infection
Long COVID symptoms change for 15 months after infection