Early on in the coronavirus disease 2019 (COVID-19) pandemic, many governments around the world enacted costly and restrictive measures to reduce the rapid transmission of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). These included mandatory face masks in public, the closing of public areas, and even full lockdowns/stay-at-home orders.
 This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Background
While the development and mass distribution of COVID-19 vaccines allowed many developed nations to begin dismantling these measures, developing nations continue to have limited access to these vaccines. Furthermore, the emergence of highly transmissible SARS-CoV-2 variants has threatened the efficacy of these vaccines as a result of their ability to evade the immune response induced by the vaccines.
Much of the scientific attention given to SARS-CoV-2 mutations is focused on mutations within the four structural proteins, particularly the spike protein, as it is key to the pathogenicity of SARS-CoV-2. The SARS-CoV-2 spike protein consists of two subunits that require cleavage by a host enzyme to become functional. These subunits include the S1 domain, which binds to the angiotensin-converting enzyme 2 (ACE2) receptor to permit viral cell entry into host cells, as well as the S2 subunit, which mediates membrane fusion.
In a recent study published on the bioRxiv* preprint server, researchers investigate SARS-CoV-2 mutations present within both the spike protein, as well as those that exist across the whole genome of different SARS-CoV-2 variants.
About the study
The researchers began by filtering all the SARS-CoV-2 genomes that were available on the GISAID database, which included over five million genomes that have been sequenced to date. This subset was further filtered using NextClade, with only good quality scores retained.
Taken together, the researchers collected about 1.7 million high-quality genomes.
The researchers then randomly selected samples and compared their genomes to selections from those obtained throughout the pandemic, including the original Wuhan reference genome and the most recent Omicron genomes.
The analysis revealed an overall increase in the number of synonymous mutations over time. Among the Omicron sublineages BA.1 and BA.2, BA.2 showed significantly more mutations than BA.1, with a more pronounced pattern observed in its number of non-synonymous mutations.
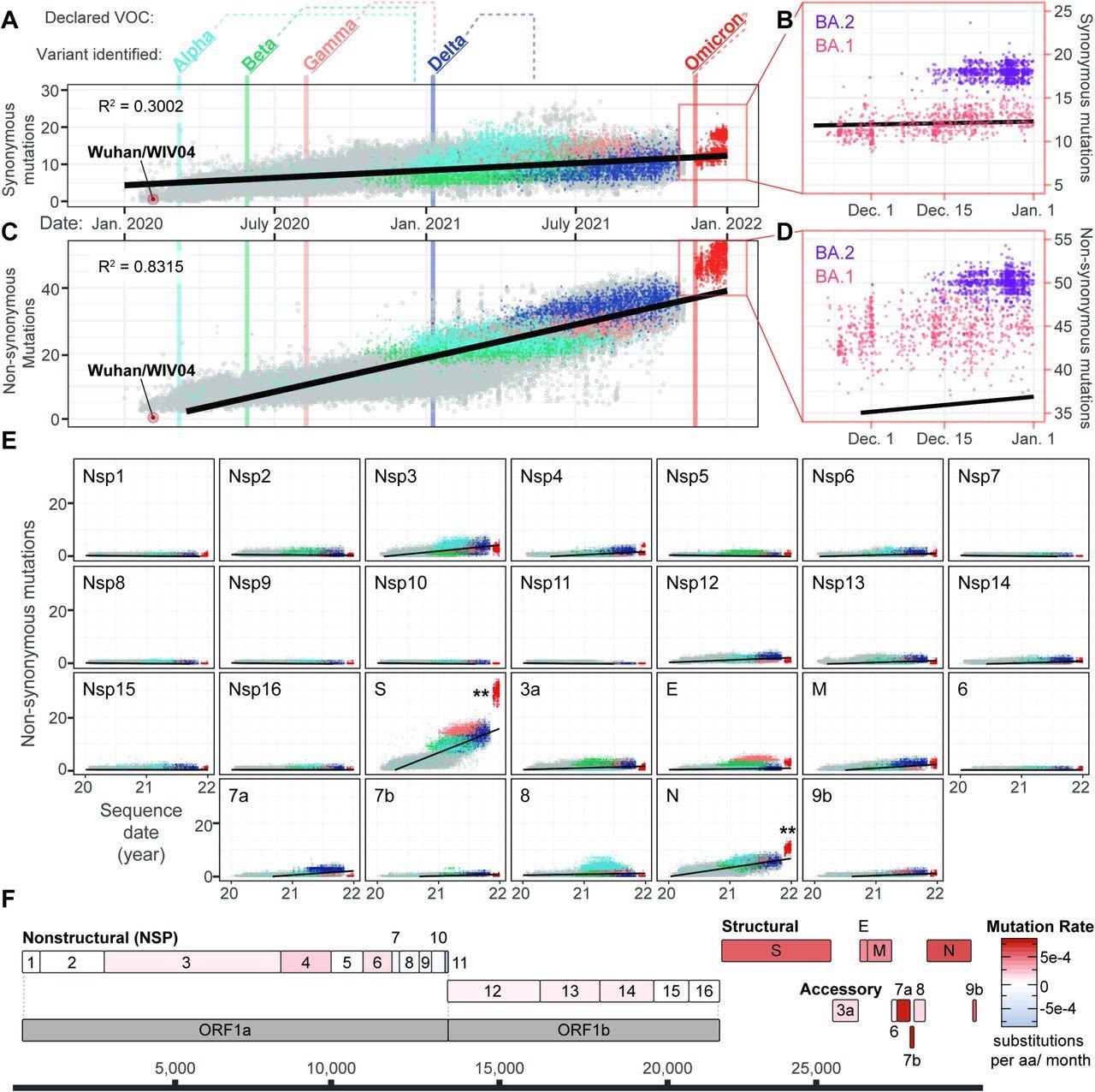
Evolution of SARS-CoV-2 and the emergence of new variants. A) Synonymous mutations in 83,204 genomes sampled from Dec 19, 2021, to Jan 14, 2022 (GISAID accessions available at: github.com/WiedenheftLab/Omicron). Variants of concern (VOC) are labeled in bold, with dates of the first sequence for each lineage shown as vertical lines through the graph. The time elapsed between first detection and VOC designation by the WHO is shown as dotted lines above the graph. Dots are colored similar to variant names, and grey circles represent non-VOC lineage genomes. A linear regression line is shown in black. Omicron genomes are <2.58 sigma from the mean, indicating that do not significantly deviate from the expected number of mutations. B) Inset from Panel A. Omicron is divided into two lineages, BA.1(red) and BA.2 (purple). Synonymous mutations are significantly different between these two lineages (T-test, p < 2e-16). C) Non-synonymous mutations in SARS-CoV-2 genomes are plotted as in panel A. Omicron variants deviate from the linear trendline (i.e., expected mutation rate) as the majority are >2.58 sigma from the mean (p < 0.01). D) Inset from Panel C. Non-synonymous in BA.1 (red) are significantly different from BA.2 (purple) (T-test, p < 2e-16). E) Non-synonymous mutations accumulated in SARS-CoV-2 proteins over time. Linear regression lines and variant colors are plotted as in panel A. Double-asterisks denote statistically significant (p < 0.01) increases in Omicron mutation rates for a given protein, compared to mutation counts predicted by linear regression analysis of non-Omicron sequences. F) Schematic depiction of SARS-CoV-2 protein-coding sequences, with each gene colored according to non-synonymous rates predicted in panel D that have been normalized to the length of each respective protein.
The scientists then analyzed each of the SARS-CoV-2 genes individually to understand which showed the greatest contribution to the significant jump in non-synonymous mutations observed in the Omicron variant. Two genes with significantly more non-synonymous mutations in both sublineages of Omicron were identified and found to encode for the spike and nucleocapsid proteins, with the spike protein of Omicron showing between 24 and 35 mutations. Notably, the Omicron nucleocapsid protein did not exhibit as many synonymous mutations; however, it had significantly more non-synonymous proteins.
As a result of their findings, the authors suggest that some COVID-19 tests and treatments that rely on the detection of the nucleocapsid protein will be less effective. It is also likely that some methods for determining whether an individual has been infected previously or has simply received the vaccine will need to change.
Previous analyses did not reveal the cause of the differences in protein length. The scientists solved this by normalizing non-synonymous mutation rates by dividing each by the number of amino acids present in the proteins.
The accessory proteins, such as 7a, 7b, and 9b, were found to change more rapidly as compared to any other viral protein. These proteins are implicated in immune suppression; therefore, these changes could partially explain why the Omicron variant is capable of escaping both naturally acquired and vaccine-induced immunity.
Conclusions
The results of the current study demonstrate that the Omicron variant shows a substantially increased rate of non-synonymous mutations as compared to previous variants and that this pattern is even more exaggerated in the BA.2 sublineage.
This news article was a review of a preliminary scientific report that had not undergone peer-review at the time of publication. Since its initial publication, the scientific report has now been peer reviewed and accepted for publication in a Scientific Journal. Links to the preliminary and peer-reviewed reports are available in the Sources section at the bottom of this article. View Sources
Journal references:
- Preliminary scientific report.
Wiegand, T., McVey, A., Nemudraia, A., et al. (2022). The rise and fall of SARS-CoV-2 variants and the emergence of competing Omicron lineages. bioRxiv. https://www.biorxiv.org/content/10.1101/2022.02.09.479842v1
- Peer reviewed and published scientific report.
Wiegand, Tanner, Artem Nemudryi, Anna Nemudraia, Aidan McVey, Agusta Little, David N. Taylor, Seth T. Walk, and Blake Wiedenheft. 2022. “The Rise and Fall of SARS-CoV-2 Variants and Ongoing Diversification of Omicron.” Viruses 14 (9): 2009. https://doi.org/10.3390/v14092009. https://www.mdpi.com/1999-4915/14/9/2009.
 Unveiling Hidden Potential: Organoids for Disease Modeling in Neuroscience Research
Unveiling Hidden Potential: Organoids for Disease Modeling in Neuroscience Research