Over the past several decades, the human microbiome has been the focus of considerable research throughout the world. Several large national and international initiatives, including the Human Microbiome Project (HMP) and MetaHIT, for example, have been conducted to better understand the complexity of the microbiome.
What causes differences in the core microbiome?
Across different geographies and populations, the core human microbiome varies greatly. These differences can be attributed to varying environmental and individual conditions such as diet, host genetics, and various other factors.
Additional differences in the core microbiome can be identified within one individual, as the gut microbiome has a distinct composition as compared to the vaginal and oral microbiomes.
Furthermore, different parts of the gut may have different microenvironments that support a distinct core microbiome. For example, mucosal-associated microorganisms have more profound effects on immune and health indices than luminal and fecal germs.
As compared to mice, gorillas, and chickens, the core human microbiome has a more common abundance of specific species. Furthermore, as compared to rodents and birds, human microbiomes are most analogous to those of gorillas.
Distinct differences also exist between Westernized and non-Westernized populations. In fact, studies have shown that several microbial species abundantly present in one human population may not ubiquitous in other human cohorts. However, Faecalibacterium prausnitzii has frequently been identified in more than 90% of the specimens within six human cohorts.
Approaches to understanding the core microbiome
The human body houses many different types of microorganisms, including commensals and pathogens. The term "core human microbiome" describes the microbiome components that remain relatively constant across time and amongst individuals.
There are primarily four different approaches for determining the core human microbiome, the two most common of which include community composition and functional profile approaches. The community composition approach describes the core microbiome in terms of the common taxa, whereas the functional profile description is based on a group of shared functions.
The ecology method defines the core microbiome based on taxon abundance, interactions, co-occurrence, and other community-level patterns. The stability approach considers characteristics that support community stability and resilience.
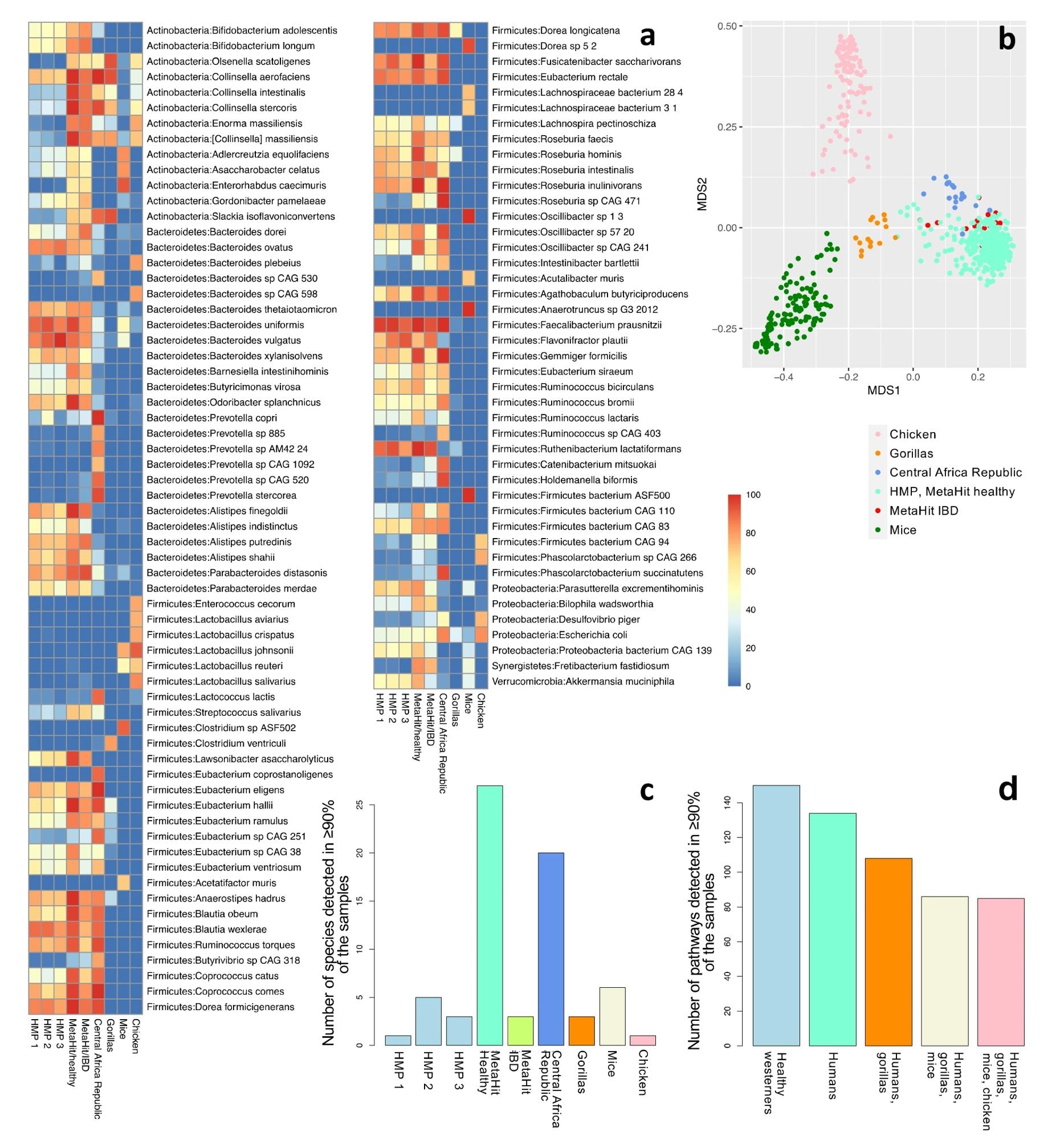
Community profiling of nine cohorts: HMP phases 1 (n = 138), 2 (n = 91), and 3 (n = 42); healthy individuals from Denmark (n = 64); individuals with IBD from Spain (n = 16); hunter-gatherers and traditional agriculturalists (n = 19); gorillas (n = 15); mice (n = 141); and chickens (n = 121). (a) The fraction of samples that contain each species for species detected in at least 70% of the samples in at least one cohort (a total of 107 species). Refer to Supplementary Table S3 for a complete list of all the detected species in all cohorts. (b) The first two components of an unweighted UniFrac-based MDS analysis considering all samples. (c) The number of species detected in at least 90% of the samples of each cohort. Only one species (F. prausnitzii) was detected in 90% or more of the samples across all healthy human Western (n = 4) and all human (n = 6) datasets. (d) The number of pathways detected in >90% of the samples in all healthy Western human datasets (n = 4); all human datasets (n = 6); human and gorilla datasets (n = 7); human, gorilla, and mouse datasets (n = 8); and all datasets, including chickens (n = 9).
The omics of the core microbiome
Currently, most research into the fundamental human microbiome relies on next-generation sequencing (NGS)-based technologies, which, when coupled with appropriate bioinformatics methodologies, create enormous volumes of data. Meanwhile, the taxonomic and functional resolution at the species level is only achievable with metagenomics, thus making it the preferred method as compared to amplicon surveys.
A considerable proportion of available data has been gathered by amplicon-based community surveys, which seek a conserved region of the target population's genomes using polymerase chain reaction (PCR) assay and specific primers.
In recent years, microbiome research has shifted its focus to amplicon-based community surveys and metagenomics, the latter of which is a method of studying microbial communities without culturing. Metagenomics relies on "shotgun" sequencing of potentially all microbial genomes present in a sample.
The European Nucleotide Archive (ENA) has over 610,000 samples of the human microbiome, 85% of which are 16S ribosomal ribonucleic acid (rRNA) amplicons. Notably, in microbiome surveys, different sites of the 16S rRNA gene are commonly used to detect prokaryotes. In conjunction with additional samples that are accessible through other platforms, ENA samples provide a sufficient amount of data for evaluating the core human microbiome.
Technological challenges
Future approaches will warrant resolution of inadequate samples from non-Western populations, as well as the lack of complete reference databases and data on eukaryote abundance, eukaryotic viruses, and phages. Other technological hurdles, such as directly sampling from different sections of the gut, must also be simplified in the future.
Therefore, the capacity to precisely assess the core microbiome of distinct areas within the gastrointestinal tract is likely to remain postponed for the foreseeable future due to the persistent scarcity of highly localized data.
In terms of metabolomics, proton nuclear magnetic resonance (1H NMR), one of the primary analytical methods utilized in these investigations, has low sensitivity and may not detect low-abundance metabolites.
In addition to NMR, both gas and liquid high-performance chromatographic techniques coupled with quadrupole-TOF (time of flight) mass spectrometry have been successfully employed to describe a large number of metabolites from microbial samples. In contrast, TOF-based detection necessitates elaborate sample pretreatment and chromatographic fractionation techniques before yielding a suitable resolution in discovery-based metabolomics.
To overcome these limitations, new high-resolution accurate-mass (HRAM) analytical tools have been utilized to study complex microbial community metabolomics. The untargeted discovery of new possible metabolites has been made possible by ultra-high performance liquid chromatography (UHPLC) coupled with a quadrupole-orbitrap mass spectrometer.
An advanced platform utilized in discovery-based proteomics and metabolomics known as the orbitrap mass analyzer provides essential tools in tackling the grey areas in host-microbiome metabolomics, despite being an expensive instrument that demands complicated data analysis abilities.
Machine learning and cloud computation
In the context of the core microbiome, the complete picture remains evasive because there is a lack of a high-throughput method capable of delivering essential information. Computational and statistical methods are required to merge data layers and uncover complicated patterns as more and more non-genomic data is collected.
To elucidate such patterns, machine learning and cloud computing are promising areas of research. Cloud computing and other high-throughput computational technologies make it possible to examine vast quantities of data, possibly including all the data held in public databases.
Conclusions
As 16S rRNA gene assessments continue to play an important role in this field, researchers will be able to make more accurate assessments of the core human microbiome using metagenomics with the aid of improved bioinformatics and data.
Taken together, experts in nutrition, immunology, human genetics, microbiome, bioinformatics, and machine learning can collaborate to develop comprehensive studies to reveal the patterns and processes that contribute to the core human microbiome.
Journal reference:
- Sharon, I., Quijada, N., Pasolli, E., et al. (2022). The Core Human Microbiome: Does It Exist and How Can We Find It? A Critical Review of the Concept. Nutrients. doi:10.3390/nu14142872
 Clear aligners protect oral microbiome better than fixed braces
Clear aligners protect oral microbiome better than fixed braces