Although coronavirus disease 2019 (COVID-19) vaccines managed to slow down the rapid transmission and severity of SARS-CoV-2 infections worldwide, the increasing number of breakthrough infections indicates the growing ability of emergent SARS-CoV-2 variants to escape the immune responses induced by vaccinations and previous infections.
Studies using neutralizing antibodies from COVID-19 patients have identified the spike protein’s receptor binding domain (RBD) and N-terminal domain (NTD) as the main targets of neutralizing antibodies. The NTD, the antigenic supersite targeted by numerous neutralizing antibodies, has also been found to carry deletions, with four regions having recurrent deletions. Since studies have shown that such deletion mutations reduce the neutralizing efficacy of neutralizing antibodies that target the NTD, it is essential to understand the role these deletions might also play in increasing the transmission abilities of SARS-CoV-2.
About the study
In the present study, the researchers used the whole genome sequences deposited in the Global Initiative on Sharing Avian Influenza Data (GISAID) database from across the world, which were approximately 2.3 million in total, as well as SARS-CoV-2 whole genome sequences generated from 102 patients with breakthrough SARS-CoV-2 infections. Epidemiological data consisting of SARS-CoV-2 positivity rates were acquired from Our World in Data (OWID) and other databases.
The monthly prevalence of mutations and SARS-CoV-2 test positivity rates were assessed across three-month intervals to determine mutations associated with sudden increases in COVID-19 cases. Mutations that showed a monotonic increase in prevalence corresponding to monotonic increases in SARS-CoV-2 positive tests were considered surge-associated mutations.
The mutations identified as associated with surges in COVID-19 cases were then compared against mutations in four variants of concern and seven variants of interest identified by the United States (U.S.) Centers for Disease Control and Prevention (CDC). The variants of concern consisted of Alpha, Beta, Delta, and Gamma variants, while the variants of interest comprised Epsilon, Eta, Iota, Kappa, and Zeta subvariants.
Additionally, the various mutation types, such as insertions, deletions, and substitutions, were assessed to determine their enrichment in surge-associated mutations. Recurrent deletion sites in the spike protein NTD were also identified. The researchers also constructed a time series tile plot to examine the temporal expansion of regions with recurrent deletions.
Results
The results reported 1045 spike protein amino acid mutations spanning insertions, deletions, and missense mutations that were present in a minimum of 100 sequences in the GISAID database. Substitution mutations comprised a large percentage, accounting for 95.21% (995) of these mutations. Deletions and insertions were 4.3% (45) and 0.48% (5), respectively. The number of surge-associated mutations — those which monotonically increased along with a monotonic increase in positive SARS-CoV-2 tests during three-month intervals — was 92, of which 42 were also found in the CDC-identified variants of interest or concern.
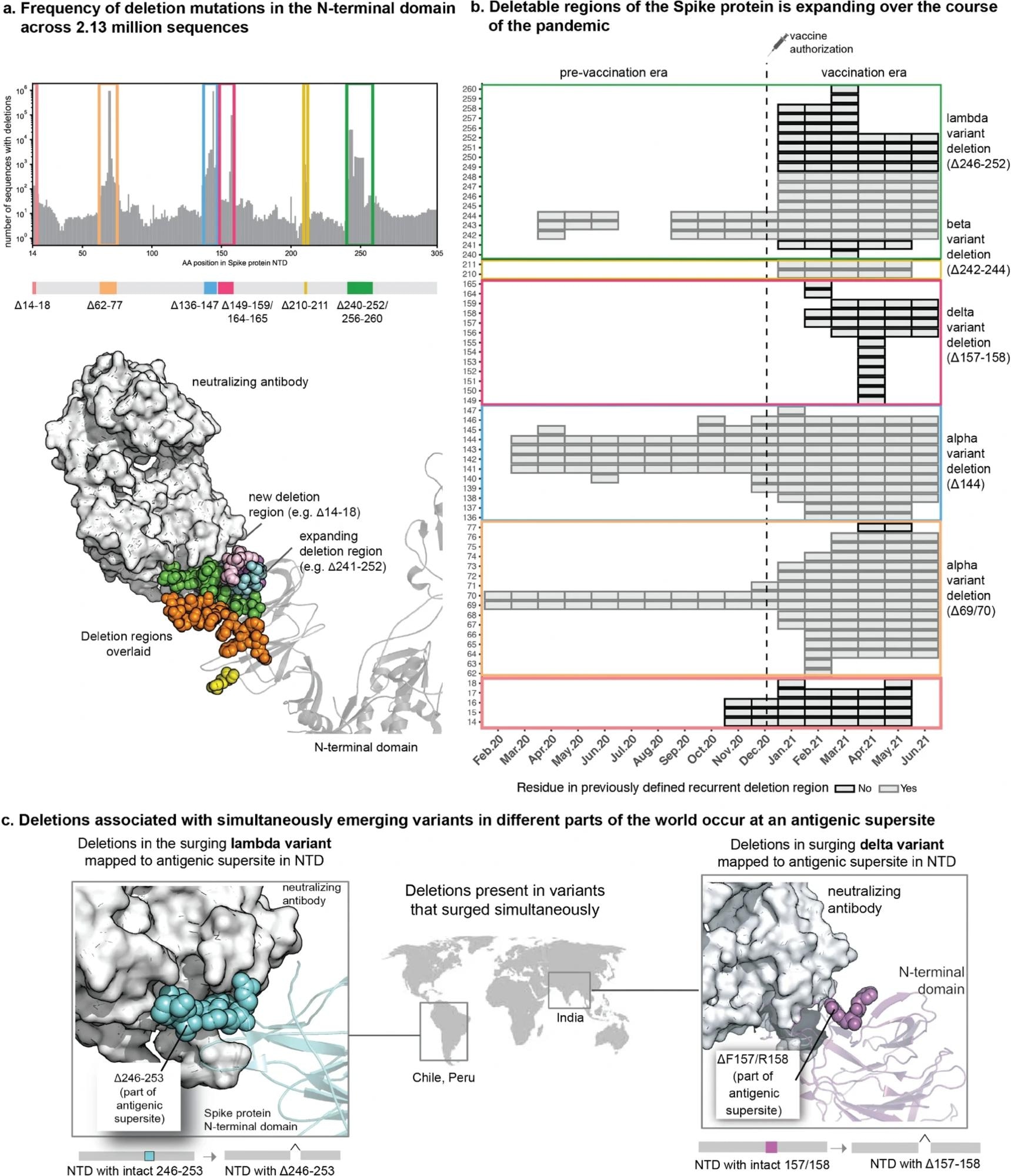 The repertoire of deletions in the Spike protein N-terminal domain is expanding over the course of the pandemic. (a) Frequency of occurrences of deletion mutations in the N-terminal domain across 2.13 million Spike protein sequences (as of 30 June 2021). The recurrent deletion regions, both known and new, are illustrated schematically and mapped on the structure of the Spike protein. Positions corresponding to the deletion mutations in the Spike protein are shown as colored spheres, and the neutralizing antibody is shown using a surface representation in grey. (b) Heatmap showing the expansion of “deletable” regions in the course of the pandemic, where the rows denote residue positions in the Spike protein and columns denote the time course of the pandemic (in months). Each box denotes the frequency of a given deletion mutation across the world in that month. The color of the boxes corresponds to a frequency of 1 to 100,000 sequences shown on a log10 scale. (c) Sites of deletion mutations associated with surges in different parts of the world are shown as spheres on the 3D structure of the Spike protein complexed with neutralizing antibody 4A8 (PDB identifier: 7C2L described by Chi et al., retrieved from the PDB).
The repertoire of deletions in the Spike protein N-terminal domain is expanding over the course of the pandemic. (a) Frequency of occurrences of deletion mutations in the N-terminal domain across 2.13 million Spike protein sequences (as of 30 June 2021). The recurrent deletion regions, both known and new, are illustrated schematically and mapped on the structure of the Spike protein. Positions corresponding to the deletion mutations in the Spike protein are shown as colored spheres, and the neutralizing antibody is shown using a surface representation in grey. (b) Heatmap showing the expansion of “deletable” regions in the course of the pandemic, where the rows denote residue positions in the Spike protein and columns denote the time course of the pandemic (in months). Each box denotes the frequency of a given deletion mutation across the world in that month. The color of the boxes corresponds to a frequency of 1 to 100,000 sequences shown on a log10 scale. (c) Sites of deletion mutations associated with surges in different parts of the world are shown as spheres on the 3D structure of the Spike protein complexed with neutralizing antibody 4A8 (PDB identifier: 7C2L described by Chi et al., retrieved from the PDB).
When surge-associated mutations were examined by mutation type, deletions were found to be associated with infection surges in greater frequencies than those expected by chance. Surges were linked with 40% of the identified deletion mutations, compared to non-deletion mutations, which only accounted for 12% of all non-deletion mutations identified in the SARS-CoV-2 genome. These deletions were also found only in the NTD region of the spike protein, suggesting a correlation between NTD-associated deletion mutations and increased transmission of the virus.
The temporal assessment of the prevalence of deletion mutations also indicated an expansion of regions containing deletion mutations surrounding the antigenic site in the NTD, which could contribute to the evolution of variants with immune evasion and increased transmission. Annotated whole genome sequences of SARS-CoV-2 from patients with breakthrough infections revealed 107 unique mutations comprising 29 deletions and 78 substitutions. Deletions between the 85th and 90th residues in the B cell epitope region were identified in five patients and are thought to have arisen since December 2020.
Conclusions
Overall, the results suggested that expanding regions and increasing frequencies of deletions, particularly in the antigenic regions surrounding the spike protein NTD, could be driving the evolution of SARS-CoV-2 variants with increased transmission and immune evasive abilities.
Journal reference:
- Venkatakrishnan, A. J., Anand, P., Lenehan, P. J., Ghosh, P., Suratekar, R., Silvert, E., Pawlowski, C., Siroha, A., Chowdhury, D. R., O’Horo, J. C., Yao, J. D., Pritt, B. S., Norgan, A. P., Hurt, R. T., Badley, A. D., Halamka, J., & Soundararajan, V. (2023). Expanding repertoire of SARS-CoV-2 deletion mutations contributes to evolution of highly transmissible variants. Scientific Reports, 13(1). https://doi.org/10.1038/s41598-022-26646-5, https://www.nature.com/articles/s41598-022-26646-5
 Azelastine nasal spray prevents COVID-19 and rhinovirus infections
Azelastine nasal spray prevents COVID-19 and rhinovirus infections