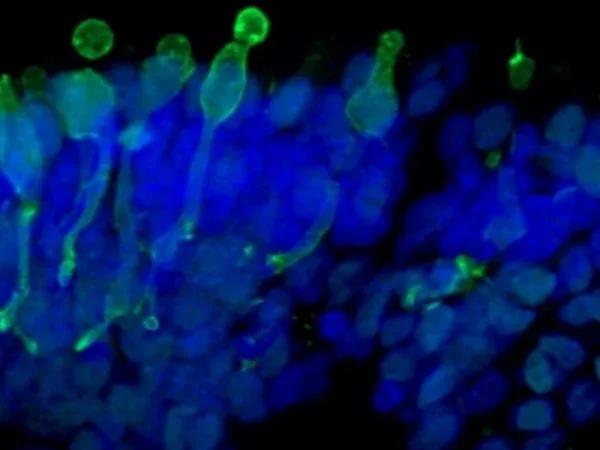
Cone photoreceptor cells labeled with anti-Opsin (Red/Green) antibody. Image Credit: Newcells Biotech
Newcells provides a reliable and quick in vitro safety and efficacy service for evaluating novel compounds for retinal therapy with complex human retinal organoid or retinal pigment epithelium (RPE) models developed in-house.
Both models are iPSC-derived from Newcells' WT line or the customer's CRISPR/Cas9 gene-edited lines, allowing for direct comparison of WT and mutant phenotypes following differentiation. Targeted mutations can simulate retinopathies, particularly monogenic inherited retinopathies, including Leber congenital amaurosis, retinitis pigmentosa (RP), Stargardt disease, and Usher’s syndrome.
Service outputs
- Photoreceptor degeneration
- Phagocytosis of photoreceptor outer segment (RPE)
- Transmission electron microscopy (TEM) and scanning electron microscopy (SEM)
- Cell viability assay
- Key markers analysis
- Qualitative immunofluorescence
- Gene expression
Models
- Human retinal organoids
- Retinal pigment epithelium (RPE)
- Derived from patients or gene-edited PBMCs or fibroblasts provided by the client
- Derived from patients or gene-edited iPSCs provided by the client
- Derived from healthy donors’ iPSCs
Timeline
How to access the retinal disease modeling service?
Outsource the experiments to obtain reliable data
Newcells designs experiments to address customer inquiries while providing a quick and dependable service. The data at the end of the study will provide important insights into the disease basis, confirming the effectiveness and safety of innovative treatments and potential rescue plans. A group of retina specialists performs the custom projects in cutting-edge UK facilities.
Generation of patient-derived functional retinal tissue as part of the disease modeling service
Take advantage of a full range of services, including creating organoids and RPE from fibroblasts, PBMCs, or patient-derived iPSCs. Newcells also offers feasibility studies, experimental design, testing of therapeutic compounds, reliable data for regulatory submissions, phenotypic rescue experiments, and comparisons between WT and patient-derived or mutated retinal tissue.
Example 1: Phenotypic analysis of Retinitis Pigmentosa mutation
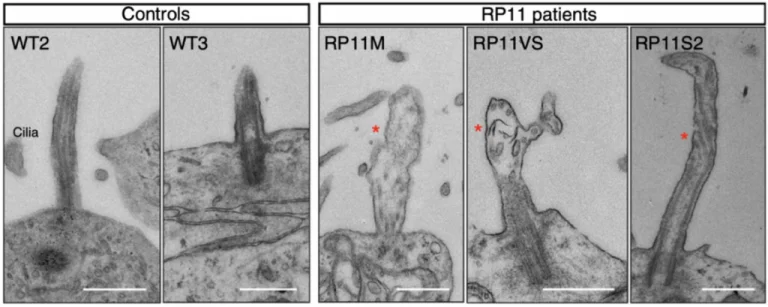
Transmission electron microscopy images showing shorter cilia in patient-derived photoreceptors, with abnormal bulbous morphology (red star). Scale bar (500 nm). Image Credit: Newcells Biotech
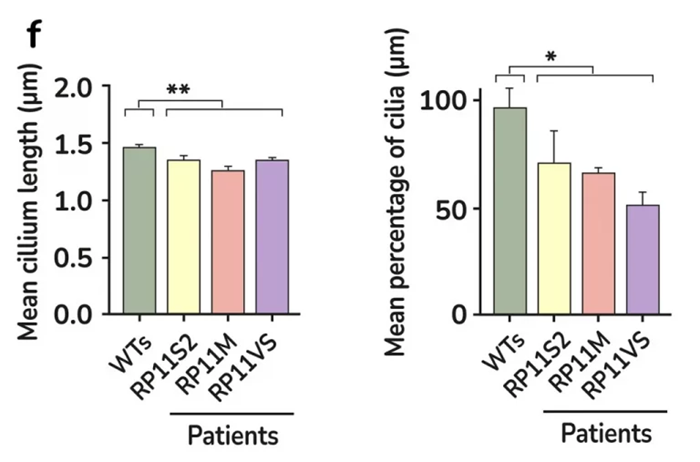
Quantification of cilia length and frequency in photoreceptors showing significant reduction in RP11 patients compared to the controls. Image Credit: Newcells Biotech
After creating retinal organoids from RP Type 11 patients with a mutation in the pre-mRNA processing factor 31 (PRPF31), the mechanism behind the retinal dysfunction in this disease has been clarified.
Large-scale transcriptome analyses that revealed patient-specific and cell type-specific mis-splicing of PRPF31 target genes impacted by PRPF31 mutations made unprecedented molecular characterization of splicing-factor RP clinical phenotypes possible.
This study uncovered several cellular defects, including progressive cellular degeneration, altered cilia morphology in photoreceptors, dysfunctional RPE, and cellular stress, as demonstrated by phenotypic rescue. The study was the first to use patient-derived organoids, or RPEs, to demonstrate the cellular phenotypes linked to RP.
This research and expertise were developed in the lab of Prof. Lako, a co-founder of Newcells Biotech and a professor of stem cell sciences at Newcastle University’s Biosciences Institute, Faculty of Medical Sciences.
Accelerate retinal therapy development
iPSC technology provides an opportunity to study disease mechanisms and assess new treatments by engineering patient-specific retinal organoids and RPE. Human iPSCs are the source of Newcells' models, creating patient-specific gene-edited lines that directly compare WT and mutant cells.
Retinal organoids are created through a meticulously regulated differentiation process that mimics the timeline of embryonic retinogenesis. At day 150, the organoids are functional and comprise all major cell types, so new compounds can be tested by adding them to the plate.
Newcells has assessed the integrity of cell structure and gene expression of key markers for major cell types, like photoreceptors, while using qualitative imaging and microscopy to get a complete view of the drug profile.
The parental iPSC line can create RPE and retinal organoids, enabling complementary retinal tissue and epithelium evaluation.
A primary advantage of these models for disease modeling is their human origin, which enables the generation of solid, predictive data for clinical and transitional studies.
Example 2: Retinal disease modeling in vitro: Age-Related Macular Degeneration
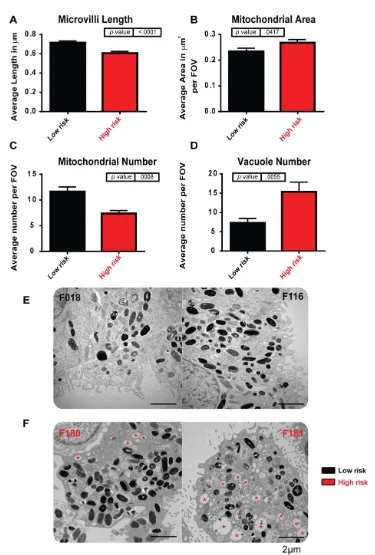
iPSC-derived retinal pigment epithelium (RPE) modeling AMD. iPSC-derived RPE generated from high-risk Y402H AMD donors (F180 and F181) show ultrastructural changes when compared to low-risk Y402H donors (F018 and F116). (A): Microvilli length is decreased in high-risk donor RPE. (B): Mitochondrial area was increased in high-risk donor RPE. (C): Mitochondrial number was decreased in high-risk donor RPE. (D): The number of vacuole structures was greatly increased in high-risk donor RPE. (E): Examples of low-risk iPSC-RPE cells: left-hand side, F018; right-hand side, F116; (F): Examples of high-risk iPSC-RPE cells: left-hand side, F180; right-hand side, F181; red asterisk indicates vacuoles. Image Credit: Newcells Biotech
The mechanisms underlying age-related macular degeneration (AMD) are poorly understood, making RPE and patient-derived retinal organoids extremely useful for disease modeling. Although the ARMS/HTRA and complement genes are known to raise the risk of AMD, it is still unclear how the other genes contribute to this increased risk.
Newcells have produced iPSC-derived RPE to model AMD from people with low- and high-risk CFH (Y402H) complement polymorphisms. In comparison to low-risk individuals, the high-risk iPSC-derived RPE cells exhibit traits typical of AMD, such as cellular, structural, and functional deficiencies linked to inflammation, cellular stress, and the buildup of lipid droplets and deposits that resemble AMD. Both the companion RPE and retinal organoids can be used for these investigations.
Service overview
Newcells' service offers insights into the molecular basis of hereditary retinopathies.
The effectiveness of new treatments can also be evaluated in vitro to speed up lead development or model phenotypic rescue. It is also possible to access and compare the safety of new drugs or viral vectors on retinal tissues derived from healthy individuals and patients.
Newcells offers a fully customized service that generates patient-specific lines or works with existing mutated iPSC lines if available. These lines are then differentiated into organoids, and a feasibility study is conducted first for incoming lines.
The differentiation protocol mimics embryonic retinogenesis and lasts 150–210 days. The project timelines are short as a regular supply of tissue is released due to the streamlined manufacturing process. When making important decisions during the drug development process, users will be confidently guided by the solid data produced by the scientific experts.
A collection of tests to evaluate cell viability, photoreceptor or RPE functionality and degeneration, and key marker expression and localization are examples of disease modeling packages. The safety of novel chemical drugs or viral vectors like AAV can also be assessed.
Source: Newcells Biotech
| Assay Design |
| Models |
Retinal organoids with photoreceptors (cone and rod), retinal ganglion cells, horizontal cells, and amacrine cells (derived from healthy (WT) or gene-edited patient-specific iPSCs provided by the client). |
| Retinal pigment epithelium (RPE) cells (2D cobblestones monolayer) (derived from healthy (WT) or gene-edited patient-specific iPSCs supplier by the client). |
| Assay format |
96-well plates (retinal organoids) Assay format 24-well plates (RPE) |
| Species |
Human |
| Cell viability assay (ATP depletion assay, LDH release and microscopy) |
| Qualitative immunofluorescence with cell-specific markers & cell death markers |
| Assay readout |
Gene expression profile of key marker gene by RT-qPC |
| Microscopy: EM, 2D-TEM, and SEM |
| Cytokine secretion assay (with RPE only) |
| Trans-epithelial electric resistance (TEER) (with RPE only) |
| Time points and replicates |
Retinal organoids disease modeling service can be performed at any time point of differentiation up to Day 210. |
| Data points are usually performed in triplicates or quadruples with a minimum of 10 organoids |
Models to choose from for this service
Retinal organoids
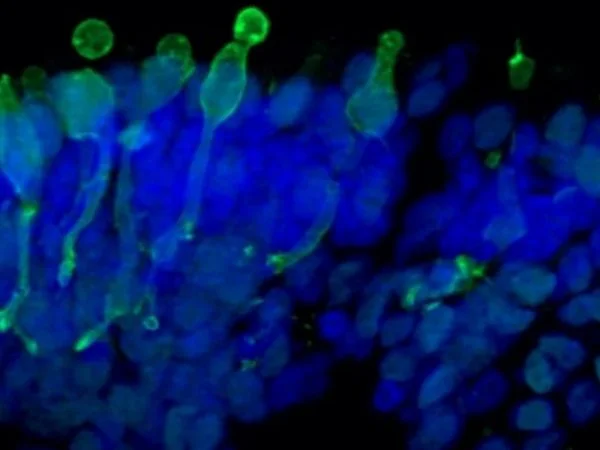
Cone photoreceptor cells labeled with anti-Opsin (Red/Green) antibody. Image Credit: Newcells Biotech
With a laminar cell organization resembling embryonic development, the retinal organoids, derived from iPSCs, replicate the intricate structure of the human retina. They contain the retina’s light-responsive outer photoreceptor segment.
Retinal pigment epithelium (RPE)
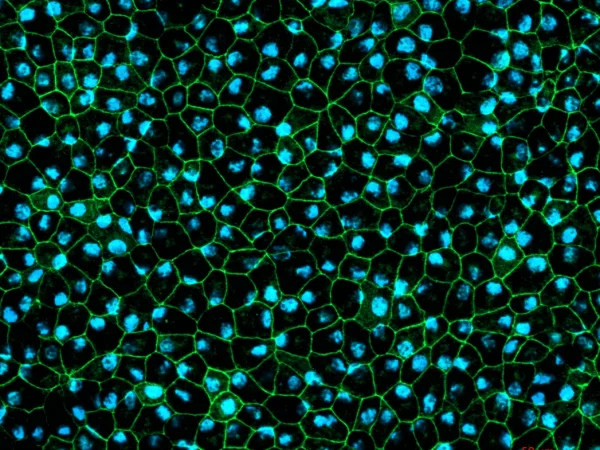
RPE cells displaying cobblestone morphology. Cells were immunolabeled with a tight-junction ZO-1 marker (shown in green) and co-stained with nuclei marker Hoechst (shown in blue). Image Credit: Newcells Biotech
A functional monolayer in vitro model of retinal pigment epithelial cells derived from human iPSCs replicates the phagocytosis of photoreceptor outer segments. The pigmented RPE cells have a cobblestone morphology.