Histamine is an aminergic neurotransmitter and plays a key role in the regulation of several pathophysiological processes. Histamine is synthesized in the mammalian brain, in restricted populations of neurons, which are found in the tuberomammillary nucleus of the posterior hypothalamus.1
These neurons diffusely project to the majority of the cerebral areas and have been implicated in many brain functions (e.g. food intake, thermoregulation, cardiovascular control, sleep/wakefulness, hormonal secretion, and memory formation).2
In peripheral tissues, histamine is stored in mast cells, basophils, eosinophils, enterochromaffin cells, and likely in some particular neurons. Mast cell histamine plays a key part in the pathogenesis of numerous allergic conditions.
After mast cell degranulation, the release of histamine results in several well-known symptoms of allergic conditions in the airway system and skin. Bovet and Staub discovered compounds in 1937, which provoke the influence of histamine on these allergic reactions.3
Since this discovery, there has been a large amount of research into finding novel ligands with (anti-) histaminergic activity. This research field has been powered by the consecutive discovery of four unique histamine receptor subtypes. Each receptor subtype has an extremely distinct pathophysiological role and they all belong to the superfamily of G-protein-coupled receptors (GPCRs).
The histamine receptor subtypes H1 and H2 have proven to be exceptional drug targets. Ligands for the histamine H3 receptor subtype are presently beginning clinical studies, and the recently discovered histamine H4 receptor subtype is the subject of huge preclinical research.
The Histamine H1 Receptor
Up until the 1970s, histamine research focused on the role of histamine in allergic diseases. This lead to the development of several potent ‘antihistamines’ (e.g. mepyramine, see figure 1), which helped combat the pronounced symptoms of allergic conditions.4
Yet, the first generation of H1 receptor antagonists, which were created for treating allergies, showed distinct side effects, for example, sedation. This specific physiological effect of the ligands was prevented by structural modifications, which stopped blood-brain-barrier penetration of the drugs.
In addition to the more recently developed antihistamines (originally termed antagonists but later reclassified as inverse agonists5), the first generation is extremely successful clinically and are widely available drugs.
The cDNA encoding a bovine H1 receptor protein was cloned in 1991, after an expression cloning strategy in Xenopus oocytes.6 The deduced amino acid sequence exhibited a 491 amino acid protein of 56 kDa.
Using the cDNA sequence encoding the bovine H1 receptor, the cDNA sequences and intronless genes encoding the human,10,11 rat,3,7 mouse,12, and guinea-pig,8,9 H1 receptor proteins were then cloned.
These receptor proteins are highly homologous, slightly different in length, and do not exhibit major pharmacological differences. Analysis of the 5 ′-flanking regions of the rat, human, and guinea pig gene7,8,10 lead to the identification of numerous DNA-binding motifs, including potential glucocorticoid responsive elements. The human H1 receptor gene resides on chromosome 3.13
The H1 receptor is part of the large family of GPCRs. The receptor is related to the phospholipase C-catalyzed formation of inositol 1,4,5-triphosphate (IP3) and 1,2-diacylglycerol (DAG). Histamine starts the generation of inositol phosphates in numerous tissues (including intestinal, brain, airway, and vascular smooth muscle24) via Gαq protein activation.14
The activation of H1 receptors can also stimulate adenylyl cyclase and the formation of cAMP in other tissues. All of the details of this signaling pathway are not understood, for example, there are still questions about the G protein and the involvement of Ca2+.15
Yet, alternative signaling pathways can be mediated by the histamine H1 receptor. Current findings show that functional heterogeneity can be ligand-directed.16 The histamine H1 receptor is an established drug target that has been studied thoroughly for decades. However, H1 receptor research continues to thrive as many new approaches and methods are being developed by utilizing this receptor as an archetypal GPCR target.
Most significantly, the histamine H1 receptor has been the subject of various molecular biology studies (e.g. large-scale overproduction17 and GPCR binding site elucidation18) over the last few years, biophysical approaches (for example solid-state NMR19) and investigations towards the general activation mechanisms of GPCRs.20,21
Ligands for H1 Receptors
The most successful approach for obtaining selective H1 agonists has been the alteration of the imidazole moiety of histamine (figure 1). The presence of the tautomeric Nπ-Nτ system of the imidazole ring is not compulsory, as shown by the selective H1 agonists 2-thiazolylethylamine and 2-pyridylethylamine. Substitution of the imidazole ring at the 2-position results in relatively selective H1 agonists.
2-(meta-halogenated) phenylhistamines, for example, are relatively potent H1 receptor agonists at the guinea-pig ileum;22 yet, these compounds behave as partial agonists in some other systems.23 A large variety of selective and potent H1 antagonists are available.4
Compounds such as triprolidine and mepyramine (also called pyrilamine) are highly potent H1 antagonists and extremely useful tools for pharmacological investigations. For example, [3H]-mepyramine, is successfully utilized as an H1 receptor radioligand.24
These so-called classical ‘antihistamines’ penetrate the brain easily and are also useful within in vivo CNS studies.2 Removal of the blood-brain barrier passage by minor structural modifications has resulted in several new, non-sedating H1 antagonists (e.g. fexofenadine, cetirizine, loratidine, and astemizole).4
Figure 1. Chemical structures of selected H1 receptor ligands. (Bold text denotes compounds available from Tocris at time of publication).
The Histamine H2 Receptor
In 1966, the pronouncement that the classical ‘antihistamines’ (i.e. H1 receptor inverse agonists) cannot antagonize all histamine-induced effects (e.g. at the heart and the stomach), led Schild and Ash to suggest two distinct subtypes of histamine receptors: H1 and H2.25
This hypothesis became widely accepted when Black et al.26 triumphed in the synthesis of a series of new compounds (e.g. cimetidine, burimamide), which could block the effects of histamine on the heart and stomach. These H2 receptor antagonists proved to be extremely helpful in the therapy of gastric ulcers.
Gantz et al.27 became the first to clone cDNA encoding a 359 amino acid H2 receptor. The H2 receptor sequence was obtained from canine gastric parietal cDNA by PCR by utilizing degenerate primers based on the known sequence similarity of various GPCRs. Subsequently, the intronless genes encoding the human,29 rats,28 guinea pig30, and mouse31 H2 receptor were cloned using homology screening.
Identification of the promoter region of the human H2 receptor gene disclosed the existence of regulatory transcription regions and sites showing inhibitory and stimulatory influences on gene expression monitored in a luciferase assay.32 Observations have shown that the human H2 receptor gene resides on chromosome 5.30
Notably, several polymorphisms have been discovered in the human H2 receptor gene33 and one of the mutations has been associated with schizophrenia.34 The histamine H2 receptor is coupled to the adenylate cyclase system in a variety of tissues (e.g. stomach, lung, gastric mucosa, heart, brain).24 Additionally, cell lines transfected with the cloned H2 receptor genes exhibited an H2 receptor-mediated increase of cAMP.35,36,37
Moreover, alternative signaling pathways for the H2 receptor have been discovered. In differentiated HL‑60 cells and CHO or HEPA cells transfected with the H2 receptor cDNA, an H2 receptor-mediated increase of the intracellular Ca2+ concentration and/or IP3 levels was seen.38,39
Furthermore, in addition to an increase in cAMP, in CHO cells expressing the rat H2 receptor, activation of the H2 receptor lead to an inhibition of the release of arachidonic acid-induced by either constitutive purinergic receptors or a Ca2+-ionophore.36 These new signal transduction pathways are both regulated via unknown, cAMP-independent pathways.
Ligands for H2 Receptors
Dimaprit was the first selective H2 receptor agonist (figure 2). This compound is nearly as active as histamine at the H2 receptor but exhibits hardly any H1 receptor activity.40 A lot later, it was revealed that dimaprit is also a moderate H3 receptor antagonist41 and a moderate H4 receptor agonist.42
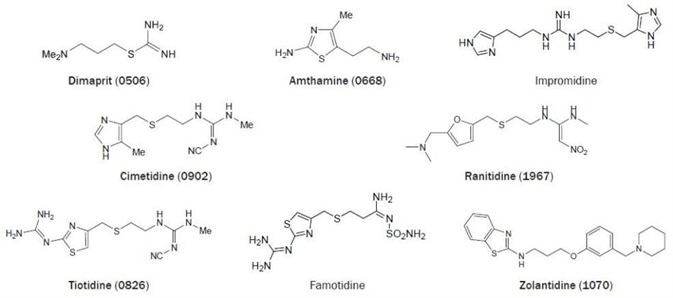
Figure 2. Chemical structures of selected H2 receptor ligands. (Bold text denotes compounds available from Tocris at time of publication).
Amthamine can be considered a rigid dimaprit analog.43 This compound combines a potency, which is slightly higher compared to histamine with a high H2 receptor selectivity, both in vivo and in vitro.44 The guanidine derivative impromidine is an H2 receptor agonist that is more potent than histamine. This ligand merges a high H2 receptor affinity with reduced efficacy.
Impromidine also exhibits moderate potent H3- and H1-receptor antagonistic activity45,46 in addition to a potent H4 receptor partial agonistic activity.42 The discovery that Nα-guanylhistamine acts as a partial H2 agonist in a gastric acid secretion test did result in the development of the relatively weak H2 antagonist burimamide.
It was shown years later that burimamide is also an H3 and H4 receptor partial agonist.42 Yet, burimamide was an excellent lead for the development of selective and clinically useful H2 receptor antagonists, like cimetidine.45,47,48
The 4-methylimidazole moiety of cimetidine can be replaced easily by other heterocyclic groups (figure 2). Replacement by a substituted furan- (e.g. ranitidine) or thiazole ring (e.g. tiotidine and famotidine) results in compounds that are typically more potent at the H2 receptor than cimetidine. Additionally, the replacement of the imidazole moiety also stops the unwanted inhibition of cytochrome P450.48
The potent tritiated H2 antagonist tiotidine and [125I]-iodinated H2 antagonist iodoaminopotentidine are utilized successfully as radioligands for the H2 receptor.24 The newly developed brain penetrating H2 antagonist zolantidine is a vital tool for in vivo CNS observations.49
The H2 receptor was noted to be spontaneously active in transfected CHO cells.50 Established using this concept, several H2 antagonists were reclassified; ranitidine, cimetidine, and famotidine are inverse agonists, yet burimamide acts as a neutral antagonist in this model system.50
The Histamine H3 Receptor
A further physiological role of histamine became apparent in the eighties, specifically its role as a neurotransmitter. In 1983, Arrang and co-workers found that the inhibitory effect of histamine on its release and synthesis was not mediated by the known H1 and H2 receptor subtypes as no correlation with either the H1 or the H2 receptor activity of known histaminergic compounds was seen.46
The H3 receptor agonist (R)-(–)-αmethylhistamine and the antagonist thioperamide (see figure 3) were developed shortly after.51 It was confirmed that this receptor subtype does regulate the release and synthesis of histamine and also has a regulatory role in the release of other neurotransmitters such as noradrenaline, serotonin, and dopamine.52
In certain regions of the CNS (for example the basal ganglia, cortical, and hippocampus areas, i.e. the parts of the brain that are associated with cognition), next to high expression, the H3 receptor is present in the peripheral nervous system, e.g. in the airways, the gastrointestinal tract, and the cardiovascular system.53,54
Utilizing the anticipated homology with the identified H1 and H2 receptor gene, initial attempts to identify the H3 receptor gene all failed. Ultimately, in 1999, the human H3 receptor cDNA was cloned by Lovenberg and co-workers.55 In search of novel GPCRs through homology searching of expressed sequence tag databases, a receptor was identified with high similarity to the M2 muscarinic acetylcholine receptor.
Expression of the gene and full characterization determined this protein as the histamine H3 receptor. The cloning of the H3 receptor of other species, including guinea pigs57, rats,56, and mice58 soon happened, and it was shown that major H3 receptor species differences exist. The human H3 receptor gene was assigned to the telomeric region of the q arm of chromosome 20 and has three exons, which are interrupted by two introns.59
The H3 receptor gene can experience extensive alternative splicing, resulting in many H3 receptor isoforms that possess different signaling properties and expression profiles.59,60,61 It was determined that the H3 receptor exhibits especially high constitutive activity, again resulting in a reclassification of existing ligands into agonists, inverse agonists and neutral antagonists.62,63
Gbahou and co-workers discovered that the compound proxyfan (figure 3) is a protean agonist both in vitro and in vivo, which means that this remarkable compound acts as an agonist, inverse agonist, or neutral antagonist, depending on the signaling pathway examined.64 The H3 receptor signals through Gi/o proteins as was demonstrated by the pertussis toxin-sensitive stimulation of [35S]-GTPγS binding in rat cortical membranes.65
Stimulation of the H3 receptor leads to reduced levels of cAMP, through negative coupling to adenylyl cyclase, and so lowering downstream signaling events like CREB-dependent gene transcription.56 Alternative signaling pathways can be activated by the Gi/o proteins, including mitogen-activated protein kinase (MAPK)60 and phosphatidylinositol 3-kinase (PI3K) pathways.
Gi/o protein activation can also result in the activation of phospholipase A2 (PLA2) to prompt the release of arachidonic acid, the inhibition of the Na+/H+ exchanger (NHE),68 and the lowering of intracellular Ca2+ levels through voltage-gated ion channels.67
With the recent progress in the characterization of the H3 receptor (as previously discussed), many pharmaceutical companies possess extremely active H3 receptor drug development programs. Several H3 receptor ligands have entered clinical studies for several applications, including dementia, narcolepsy, obesity, and migraine (to name a few) as a result.69,70
Ligands for H3 Receptors
Histamine itself is a highly active agonist at the H3 receptor. Alteration of the endogenous ligand by mono- or dimethylation of the amino function leads to compounds which are more selective and active for the H3 receptor. Methylation of the α‑carbon atom of the ethylamine sidechain dramatically increases the potency at the H3 receptor.
This increased activity resides completely in the R-isomer; the corresponding S-isomer is around 100 times less potent. (R)-(–)-αmethylhistamine (figure 3) was initially considered a selective agonist at the H3 receptor, as the methylation results in drastically reduced activity at both H1 and H2 receptors.
Yet, when the H4 receptor was uncovered it was observed that (R)-(–)-α-methylhistamine also has a sizeable affinity for this histamine receptor subtype.42 However, in combination with its less active S-isomer, (R)-(–) -α-methylhistamine has proven to be highly useful for pharmacological characterization of H3 receptor-mediated effects.41
Tritiated forms of Nα-methylhistamine and (R)-(–)-αmethylhistamine are available as radiolabelled agonists for the H3 receptor. For potent H3 agonism, the amine function of histamine can be replaced by an isothiourea group, as in imetit. This compound is also extremely active in vitro and in vivo,71,72,73 as is (R)-(–)-α-methylhistamine. The amine function can also be included in ring structures.
For example, immepip is a potent H3 receptor agonist that is useful in vitro and in vivo.74 Although the outlined first-generation H3 agonists were utilized intensively as reference ligands to study the H3 receptor, each one of them proved to also possess considerable activity at the H4 receptor.
So, a new generation of selective and potent H3 receptor agonists has been created, most significantly immethridine75 (pEC50 = 9.74; displays 300-fold selectivity over the H4 receptor) and methimepip.76 These latter compounds are lacking high H4 receptor activity. Similar to the first-generation H3 receptor agonists, the first-generation H3 receptor antagonists (all of them have an imidazole heterocycle) were discovered to have an affinity for the H4 receptor.
The first potent H3 receptor antagonist (later reclassified as an inverse agonist) that was without H1 receptor and H2 receptor activity was thioperamide (figure 3).51 This compound has been utilized in a number of H3 receptor studies as a reference ligand and is active in vitro and in vivo (the compound is able to penetrate the CNS).77
Figure 3. Chemical structures of selected H3 receptor ligands. (Bold text denotes compounds available from Tocris at time of publication).
Yet, thioperamide shows some 5-HT3 receptor antagonism78 and is an inverse agonist at the H4 receptor. The remarkable H3 receptor species difference can be shown with thioperamide, as it possesses a 10-fold higher affinity for the rat H3 receptor than for the human H3 receptor.56
Based on the H3 receptor agonist imetit (vide ante), the highly potent H3 receptor inverse agonist clobenpropit was created (pA2 = 9.9).73 This compound has some 5-HT3 receptor activity78 and shows some agonist activity at H4 receptors. Furthermore, radioligands for the H3 receptor have been described (e.g. [125I]-iodophenpropit and [125I]-iodoproxyfan).79,80
The moderately active H2 receptor antagonist burimamide (pA2 = 5.1, figure 2) also has good affinity for the H3 (pKi = 7.9) and the H4 (pKi = 7.4) receptor.42 Impentamine is a potent histamine H3 receptor inverse agonist (pA2 = 8.4). Similarly to burimamide, this compound can act as a partial agonist in SK-N-MC cells which express human H3 receptors.
Additionally, it was discovered that small structural modifications of impentamine, i.e. alkylation of the primary amine moiety of impentamine with e.g. isopropyl-, methyl-, and p-chlorobenzyl- groups leads to ligands which cover the full spectrum of functional activity, i.e. agonism, inverse agonism, and neutral antagonism.63,81
The compound VUF 5681 was found to be a neutral H3 antagonist (or ‘silent’ antagonist),82 findings which were confirmed recently in studies investigating constitutive activity utilizing rat brain cortex.83 It has been revealed that the functional activity of proxyfan relies on the system employed, ranging from inverse agonist to full agonist. Hence, proxyfan can be classified as a protean agonist, both in vitro and in vivo.64,84
In 1998, the first non-imidazole H3 receptor-ligand was reported by Ganellin.85 The potent compound UCL 2190 was a result of this elegant medicinal chemistry work.86 In 1999, after the cloning of the H3 receptor, numerous pharmaceutical companies entered the H3 research field and explored this structural class further.
JNJ 5207852 is an example of these new ligands. It is a compound that has some typical structural features for H3 receptor ligands, e.g. piperidinyl propyloxy side chain. This neutral antagonist is active in numerous models for cognition87,88 but has no effect on food intake and does not act as an appetite suppressant.89
Other compounds, like Abbott’s A 423579, seem to have more efficacy in obesity models while lacking clear cognitive effects.90 At the time of writing, the differences in efficacy for distinct clinical applications of the different classes of H3 ligands is not known and is the subject of extensive research.70,91 Interestingly, GSK 189254 has featured in trials for three different diseases: narcolepsy, neuropathic pain, and dementia.92
The Histamine H4 Receptor
Numerous groups identified the homologous H4 receptor sequence in the human genome databases straight after the cloning of the H3 receptor gene.93–97 The H4 receptor does, in fact, have high sequence identity with the H3 receptor (54% in the transmembrane domains, 31% at the protein level).
The H3 and H4 receptors are also alike in gene structure. The human H4 receptor gene is found on chromosome 18q11.2 in a single copy per haploid genome. The gene spans over 21 kbp and has three exons which are interrupted by two large introns.98 Two H4 receptor isoforms have been identified to date.
Cloning of the genes that encode the rat, mouse, guinea-pig, and pig H4 receptors only show limited sequence homology with the human H4 receptor. The H4 receptor is expressed mainly in peripheral leukocytes and bone marrow, and mRNAs of the human H4 receptor are found in dendritic cells, mast cells, eosinophils and spleen.93–97
The receptor has a noticeable influence on the chemotaxis of numerous cell types which are associated with inflammatory and immune responses. The H4 receptor is predominantly coupled to Gi/o proteins, resulting in a decrease in the production of cAMP and subsequent downstream effects such as regulation of cAMP-responsive element-binding protein (CREB) gene transcription.
H4 receptor stimulation also influences the pertussin toxin-sensitive activation of mitogen-activated protein (MAP) kinase pathways. By examining the increased levels of [35S] GTPγS levels in H4 transfected cells, it has been revealed that the H4 receptor is constitutively active. The Gβγ subunits of the Gi/o proteins activate phospholipase C, and so heighten the concentrations of Ca2+.
This Ca2+ response can be linked to cellular chemotaxis in mast cells and eosinophils. Numerous applications are under preclinical investigation considering the physiological role of the H4 receptor,99,100 including asthma and allergy,101 in addition to chronic inflammations like inflammatory bowel disease (IBD)102 and rheumatoid arthritis.103 The H4 receptor is also involved in the progression of colon cancer106 and is being associated with pruritus (itch).104,105
Figure 4. The first selective H4 receptor ligands reported in the scientific literature. (Bold text denotes compounds available from Tocris at time of publication).
Ligands for H4 Receptors
As discussed, most first-generation, imidazole-containing H3 receptor ligands also have a considerable affinity for the H4 receptor. Yet, selective H4 receptor ligands have been described.
4-Methylhistamine is a potent H4 agonist while showing more than 100-fold selectivity over the other histamine receptor subtypes, including the H2 receptor for which this ligand was originally made.42 A marginally different and complementary profile was detailed for VUF 8430. This compound has a high H4 receptor affinity and activity, a minute affinity for the H1 and H2 receptor and a 33-fold lower affinity for the H3 receptor.107
Potent and selective H4 receptor antagonists are also materializing. The first reported neutral antagonist is JNJ 7777120, which is a compound that is regularly being utilized as an H4 receptor reference ligand. Presently, [3H]-JNJ 7777120 and [3H]-histamine are employed as H4 receptor radioligands. Sadly, JNJ 7777120 has poor stability in rat and human liver microsomes and a short half-life of around two hours.
The benzimidazole derivative JNJ 10191584 (VUF 6002) is also a neutral H4 antagonist.108 This compound is orally active in vivo and has an enhanced liver microsome stability but still has a limited half-life.102,109 More recently, 2‑arylbenzimidazoles were reported as ligands with low nanomolar affinity for the H4 receptor.110
It is predicted that many new H4 receptor ligands will be described in scientific literature shortly, considering the amount of H4 receptor-related patent applications that have recently been disclosed (as reviewed elsewhere).100
Conclusions
The histamine receptor research field continues to grow as members of industry and academia find this family of receptors extremely rewarding, both commercially and scientifically. For more than seventy years, considerable efforts have been assigned to discovering new ways to modulate the different physiological processes which are mediated by histamine.
The consecutive discovery of new histamine receptor subtypes (distinction between H1 and H2 receptors in 1966, discovery of the H3 receptor in 1983 and the H4 receptor in 2000, note the interval of seventeen years) supplies an exciting and complete platform for research.
The histamine receptor family looks to also hold the promise of GPCRs as superb drug targets with two receptor subtypes (H1 and H2) addressed by blockbuster drugs, the third subtype (H3) resulting in frantic clinical studies and the latest addition to the family (H4) resulting in extremely interesting preclinical data. Therefore, it can be predicted that histamine receptor research will continue to grow in the coming years.
Special Mentions
This white paper was based on the original work of:
Iwan de Esch and Rob Leurs:
Dr. Iwan de Esch is an assistant professor and Prof. Rob Leurs is a full professor and head of the Division of Medicinal Chemistry of the Leiden/Amsterdam Center of Drug Research (LACDR), VU University Amsterdam, The Netherlands. Since the seventies, histamine receptor research has been one of the traditional themes of the division. Molecular understanding of ligand-receptor interaction is obtained by combining pharmacology (signal transduction, proliferation), molecular biology, receptor modeling and the synthesis and identification of new ligands.
References and Further Reading
- Schwartz et al. (1991) Physiol.Rev. 71 1.
- Yanai et al. (2007) Pharmacol.Ther. 113 1.
- Bovet et al. (1937) C.R.Soc.Biol.Ses.Fil. 123 547.
- Zhang et al. (1997) In: Burger’s Medicinal Chemistry and Drug Discovery, Fifth Edition. Ed. Wolff, p 495. John Wiley & Sons, Inc.
- Leurs et al. (2002) Clin.Exp.Allergy 32 489.
- Yamashita et al. (1991) Proc.Natl.Acad.Sci.USA 88 11515.
- Fujimoto et al. (1993) Biochem.Biophys.Res.Comm. 190 294.
- Horio et al. (1993) J.Biol.Chem. 114 408.
- Traiffort et al. (1994) J.Neurochem. 62 507.
- Fukui et al. (1994) Biochem.Biophys.Res.Comm. 201 894.
- De Backer et al. (1993) Biochem.Biophys.Res.Comm. 197 1601.
- Inoue et al. (1996) Genomics 36 178.
- Leconiat et al. (1994) Hum.Genet. 94 186.
- Hill et al. (1997) Pharmacol.Rev. 49 253.
- Leurs et al. (1994) J.Neurochem. 62 519.
- Noniri et al. (2004) J.Pharmacol.Exp.Ther. 311 274.
- Ratnala et al. (2004) Eur.J.Biochem. 271 2636.
- Bruysters et al. (2005) Mol.Pharmacol. 67 1045.
- Ratnala et al. (2007) J.Am.Chem.Soc. 129 867.
- Xie et al. (2006) J.Pharmacol.Exp.Ther. 317 139.
- Jongejan et al. (2005) Nat.Chem.Biol. 1 371.
- Zingel et al. (1995) In: Progress in Drug Research, Ed. Jucker, p 49, Birkhouser Verlag, Basel.
- Leurs et al. (1994) Biochem.Biophys.Res.Comm. 201 295.
- Leurs et al. (1995) Pharmacol.Ther. 66 413.
- Ash and Schild (1966) Br.J.Pharmacol. 27 427.
- Black et al. (1972) Nature 236 385.
- Gantz et al. (1991) Proc.Natl.Acad.Sci.USA 88 429.
- Ruat et al. (1991) Biochem.Biophys.Res.Comm. 179 1470.
- Gantz et al. (1991) Biochem.Biophys.Res.Comm. 178 1386.
- Traiffort et al. (1995) Biochem.Biophys.Res.Comm. 211 570.
- Kobayashi et al. (1996) Genomics 37 390.
- Nishi et al. (1995) Biochem.Biophys.Res.Comm. 210 616.
- Orange et al. (1996) Neuroreport 7 1293.
- Orange et al. (1996) Mol.Psychiatry 1 466.
- Leurs et al. (1994) Br.J.Pharmacol. 112 847.
- Traiffort et al. (1992) Proc.Natl.Acad.Sci.USA 89 2649.
- Delvalle et al. (1992) Am.J.Physiol. 263 G967.
- Mitsuhashi et al. (1989) J.Biol.Chem. 264 18356.
- Seifert et al. (1992) Mol.Pharmacol. 42 227.
- Parsons et al. (1977) Agents Actions 7 31.
- Leurs et al. (1995) In: Progress in Drug Research Ed. Jucker, p 107, Birkhauser Verlag, Basel.
- Lim et al. (2005) J.Pharmacol.Exp.Ther. 314 1310.
- Eriks et al. (1992) J.Med.Chem. 35 3239.
- Coruzzi et al. (1996) Arch.Pharmacol. 353 417.
- Durant et al. (1978) Nature 276 403.
- Arrang et al. (1983) Nature 302 832.
- Ganellin (1992) In: The Histamine Receptor. Eds. Schwartz and Haas, p 1. Wiley-Liss, New York.
- Van der Goot et al. (1991) In: Handbook of Experimental Pharmacology. Ed. Uvnas, p 573. Springer-Verlag, Berlin.
- Young et al. (1988) J.Med.Chem. 31 626.
- Smit et al. (1996) Proc.Natl.Acad.Sci.USA 93 6802.
- Arrang et al. (1987) Nature 327 117.
- Schlicker et al. (1994) Fund.Clin.Pharmacol. 8 128.
- Martinez-Mir et al. (1990) Brain Res. 526 322.
- Bertaccini et al. (1998) In: The histamine H3 receptor; a target for new drugs. Eds. Leurs and Timmerman, p 59. Elsevier
- Lovenberg et al. (1999) Mol.Pharmacol. 55 1101.
- Lovenberg et al. (2000) J.Pharmacol.Exp.Ther. 293 771.
- Tardivel-Lacombe et al. (2000) Neuroreport 11 755.
- Chen et al. (2003) Eur.J.Pharmacol. 467 57.
- Tardivel-Lacombe et al. (2001) Neuroreport 12 321.
- Drutel et al. (2001) Mol.Pharmacol. 59 1.
- Bakker et al. (2004) Inflamm.Res. 53 509.
- Morisset et al. (2000) Nature 408 860.
- Wieland et al. (2001) J.Pharmacol.Exp.Ther. 299 908.
- Gbahou et al. (2003) Proc.Natl.Acad.Sci.USA 100 11086.
- Clark and Hill (1996) Eur.J.Pharmacol. 296 223.
- Rouleau et al. (2002) Br.J.Pharmacol. 135 383.
- Silver et al. (2002) Proc.Natl.Acad.Sci.USA 99 501.
- Silver et al. (2001) Proc.Natl.Acad.Sci.USA 98 2855.
- Leurs et al. (2005) Nat.Rev.Drug Discov. 4 107.
- Celanire et al. (2005) Drug Discov.Today. 10 1613.
- Garbarg et al. (1992) J.Pharmacol.Exp.Ther. 263 304.
- Howson et al. (1992) Bioorg.Med.Chem.Lett. 2 77.
- Van der Goot et al. (1992) Eur.J.Med.Chem. 27 511.
- Vollinga et al. (1994) J.Med.Chem. 37 332.
- Kitbunnadaj et al. (2004) J.Med.Chem. 47 2414.
- Kitbunnadaj et al. (2005) J.Med.Chem. 48 2100.
- Mochizuki et al. (1996) Brain.Res. 743 178.
- Leurs et al. (1995) Br.J.Pharmacol. 116 2315.
- Ligneau et al. (1994) J.Pharmacol.Exp.Ther. 271 452.
- Jansen et al. (1994) Br.J.Pharmacol. 113 355.
- Govoni et al. (2006) J.Med.Chem. 49 2549.
- Kitbunnadaj et al. (2003) J.Med.Chem. 46 5445.
- Moreno-Delgado et al. (2006) Neuropharmacology 51 517.
- Krueger et al. (2005) J.Pharmacol.Exp.Ther. 314 271.
- Ganellin et al. (1998) Arch.Pharmacol. (Weinheim) 331 395.
- Schwartz et al. (2000) Patent WO0006254.
- Passani et al. (2004) TiPS 25 618.
- Hancock et al. (2004) Exp.Opin.Invest.Drugs 13 1237.
- Barbier et al. (2004) Br.J.Pharmacol. 143 649.
- Hancock et al. (2005) Exp.Opin.Invest.Drugs 14 223.
- Yoshimoto et al. (2006) Proc.Natl.Acad.Sci.USA 1003 13866.
- Woolley et al. (2006) Abstr. BAP Summer Meeting, Oxford, UK TE12
- Oda et al. (2000) J.Biol.Chem. 275 36781.
- Liu et al. (2001) Mol.Pharmacol. 59 420.
- Nguyen et al. (2001) Mol.Pharmacol. 59 427.
- Morse et al. (2001) J.Pharmacol.Exp.Ther. 296 1058.
- Zhu et al. (2001) Mol.Pharmacol. 59 434.
- Coge et al. (2001) Biochem.Biophys.Res.Comm. 284 301.
- De Esch et al. (2005) TiPS 26 462.
- Lim et al. (2006) Curr.Top.Med.Chem. 6 1365.
- Fung-Leung et al. (2004) Curr.Opin.Invest.Drugs 5 1174.
- Varga et al. (2005) Eur.J.Pharmacol. 522 130.
- Ikawa et al. (2005) Biol.Pharm.Bull. 28 2016.
- O’Donoghue et al. (2007) Dermatol.Ther. 18 333.
- Dunford et al. (2007) J.Allergy Clin.Immunol. 119 176.
- Cianchi et al. (2005) Clin.Cancer Res. 11 6807.
- Lim et al. (2006) J.Med.Chem. 49 6650.
- Terzioglu et al. (2004) Bioorg.Med.Chem.Lett. 14 5251.
- Venable et al. (2005) J.Med.Chem. 48 8289.
- Lee-Dutra et al. (2006) Bioorg.Med.Chem.Lett. 16 6043.
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.