In accordance with the features of RAS protein function essential for RAS-dependent cancer growth (membrane association, GTP binding, effector signaling), a variety of logical directions have been pursued to enable the development of pharmacologic strategies which can disrupt mutant RAS function in cancer (Figure 1).1,2
The most ideal strategy for targeting RAS is to produce direct inhibitors of RAS function. Although contemporary research has discovered small molecules that directly bind to and selectively unsettle the activity of mutant RAS, most of the pursued strategies are indirect, targeting proteins that support RAS function. The clear potential limit of this approach is that indirect targets are also likely to support the function of additional proteins. Therefore, indirect inhibitors of RAS will likely be compromised by off-target normal cell toxicity.
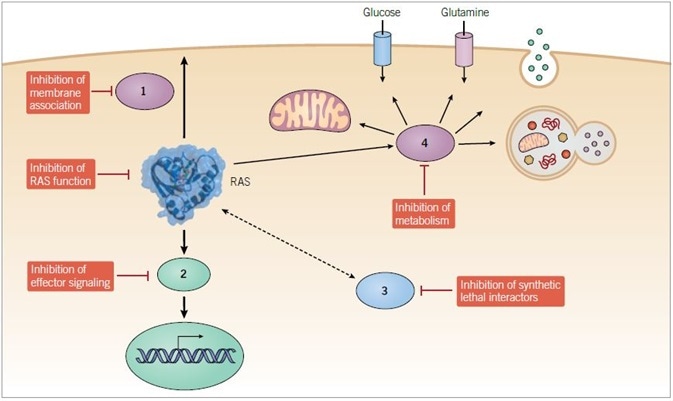
Figure 1. Anti-RAS therapeutic approaches. Direct and indirect strategies are being pursued to develop small molecule inhibitors of mutant RAS function. Indirect approaches include inhibitors of (1) proteins that facilitate RAS membrane association, (2) downstream effectors, (3) synthetic lethal interactors of mutant RAS, and (4) modulators of RAS-dependent metabolic activities.
Direct Inhibitors of RAS
Since mutant RAS proteins are GAP-insensitive and resolutely GTP-bound, appealing and logical strategies for inhibiting their function include small molecules either operating as GAP mimetics or disrupting GTP binding.1,2 Nevertheless, to date, neither technique has been effective. Although the efficacious development of ATP-competitive protein kinase inhibitors for cancer treatment (e.g., erlotinib, vemurafenib) suggests that similar techniques should be practicable with RAS, GTP binding to RAS occurs at low picomolar affinities, in distinction to the low micromolar binding affinity of ATP for protein kinases.3
In conjunction with the millimolar concentrations of GTP in cells, this high affinity means that effectual GTP-competitive small molecule inhibitors of RAS are not feasible. The crystal structure of HRAS did not manifest any well-defined pockets to reinforce the development of selective and robust small molecules that identify and disrupt the activity of mutant RAS proteins. This generated a broadly acknowledged understanding that RAS proteins are “undruggable”.
Interestingly, contemporary research with KRAS4B has started to problematize this understanding, due to the identification of a number of direct binding small molecules that are able to disrupt divergent features of RAS function.45,46 Cell-active small molecules that tether to KRAS and inhibit its interaction with the RASGEF SOS1 (DCAI, VU0460009) or its association with RAS effectors (Kobe 0065 and Kobe 2602) have been delineated. Notably, using a computational design methodology, compound 3144 underwent development as an antagonist of KRAS interaction with effectors and affects all RAS isoforms.47
Small molecules which selectively identify and inhibit the activity of one specific KRAS mutant, a G12C substitution (SML-8-73-1, ARS-853) are among the direct binders that have attracted the most substantial attention.48–51 The capability of ARS-853 to selectively block G12C function also demonstrated that at least some oncogenic mutants preserve a dependency on GEF activity to attain the completely activated state. Therefore, molecules that inhibit GEF stimulation might be effectual among a subset of KRAS mutants.
The non-steroidal anti-inflammatory drug sulindac, as well as its derivatives, have exhibited anticancer activity, and this activity can be partially mediated by direct binding to RAS and by blocking RAS interaction with RAF.52–55 However, sulindac inhibition of RAF signaling might be indirect.56 It remains to be seen whether the antitumor behavior of sulindac and its derivatives are as a result of direct interaction with RAS and impairment of effector activation.
Inhibitors of RAS Membrane Association
Since RAS plasma membrane association is critical for RAS oncogenic operation, the development of pharmacologic techniques which can target the relevant enzymes has also been an appealing strategy for antiRAS drug discovery.29,41
The discovery of FTase in 1990 instigated a robust attempt by several ceutical companies to bring through FTase inhibitors (FTIs). FTIs exhibited excellent anti-tumor activity in multiple cell culture and mouse models of RAS-driven cancers, with two FTIs (tipifarnib, lonafarnib) progressing to phase III clinical evaluation. Unfortunately, FTIs demonstrated no clinical effectiveness when assessed in cancers with a high frequency of KRAS mutations. The basis for this had been exposed years before when it was established that FTIs were not effectual inhibitors of NRAS or KRAS membrane association.
In contrast to HRAS, the isoform most broadly examined in the initial years of anti-RAS drug discovery, the NRAS and KRAS isoforms most regularly mutated in cancer are subject to alternative prenylation in FTI-treated cells. While they are not usually substrates for the related enzyme geranylgeranyltransferase-I (GGTase-I), when FTase activity is inhibited, both NRAS and KRAS are altered by this enzyme via covalent addition of a C20 geranylgeranyl isoprenoid lipid, thus restoring RAS membrane association.
Despite the failure of FTIs as an effectual treatment for cancers in which NRAS and KRAS mutations are predominant, there has been a recent reconsideration of FTIs for therapy of cancers harboring HRAS mutations. An obvious solution for the limits of FTIs would be to integrate them with a GGTI inhibitor (e.g., GGTI-2417). However, since these two enzymes collectively possess hundreds of CAAX motif-terminating protein substrates, off-target toxicity is a possible issue. Nonetheless, the possibility of parallel inhibition of GGTase-I and FTase has not yet been robustly analyzed.57
Inhibitors of additional proteins, which enable RAS membrane association, have also undergone development. Salirasib (S-farnesylthiosalicylic acid, FTS) is a further small molecule designed as an antagonist for RAS membrane association.58 Developed as a mimetic of the carboxyl-terminal farnesylated cysteine modification, cell culture research has shown various mechanisms by which salirasib can inhibit RAS function. These comprise decreased RAS protein stability, decreased RAS association with the plasma membrane, and modified effector interactions.59
Work targeting the additional enzymes implicated in CAAX-signaled posttranslational modifications has also been undertaken, but with limited success. No inhibitors with adequate selectivity and potency have undergone development, although relatively nonspecific pharmacologic inhibitors of RCE1 and ICMT have been delineated. The relatively nonspecific PAT inhibitor 2-bromopalmitate (2-BP) is a tool compound that inhibits the plasma membrane association of palmitoylated RAS isoforms. Phosphodiesterase 6 delta (PDEd), a chaperone protein that enables the trafficking of RAS proteins from endomembranes to the plasma membrane, represents one potentially promising target.60,61 Structurally distinct small molecules (e.g., Deltazinone 1, Deltarasin, and Deltasonamide 1/2),60,62 that disrupt the capacity of PDEd to engage KRAS have been proven to lower KRAS plasma membrane association and to exhibit anti-tumor behavior in vitro and in vivo.
In recent years, unbiased screens have been implemented to detect novel components that modulate RAS membrane interaction. One research program determined the L-type calcium channel blocker fendiline to be a specific inhibitor of KRAS4B but not NRAS or HRAS plasma membrane association, in a manner not related to calcium channels.63,64
Fendiline inhibition of acid sphingomyelinase and depletion of plasma membrane phosphatidylserine and cholesterol were established as the basis for selective disruption of KRAS4B membrane association. To summarize, although still an active research focus, many components which impact on RAS subcellular trafficking and membrane interactions also modulate the functions of a number of additional proteins. Nonetheless, although there is a persistent concern that inhibitors of these components might be restricted by off-target activities, it is also feasible that off-target functions of other agents may be implemented to inhibit RAS.
Inhibitors of RAS Effector Signaling
Due to the fact that protein kinases are features of effector signaling and drug targets embodying high tractability, inhibitors of specific components of RAS downstream effector signaling comprise one of the more energetic sectors in small molecule development for anti-RAS drug discovery.1,33 Of these, the principal research focus has been on RAF-MEK-ERK MAPK signaling, wherein the regular mutational activation of BRAF confirms the driver role of this pathway in cancer. Point mutations of activated BRAF alleles are in a considerable subset of the rare (<5%) PDACs lacking mutant RAS alleles.19,65,66 Among mouse models of PDAC, mutant Braf phenocopied mutant Kras to drive the initiation and development of metastatic PDAC.67 In this way, the ERK MAPK cascade is evidently a critical driver of KRAS-mutant PDAC development.
A number of potent and selective inhibitors of each stage in the RAFMEK-ERK mitogen-activated protein kinase cascade have undergone development, with inhibitors of MEK (trametinib, cobimetinib) and RAF (vemurafenib, dabrafenib) receiving approval for treating BRAF-mutant melanoma.33,68 Unfortunately, the first-generation RAF inhibitors are BRAF-selective, causing paradoxical activation instead of inactivation of ERK signaling. Pan-RAF (LY3009120) or “paradox breaker” inhibitors, which have recently undergone development, might overcome this problem.69,70
In more recent years, since resistance mechanisms overcoming sensitivity to MEK and RAF inhibitors engender ERK reactivation, a number of ERK-selective inhibitors have undergone development (VTX11e, SCH772984), exhibiting the capacity to overcome these resistance mechanisms.71 At present, three further ERK inhibitors are undergoing clinical evaluation (LTT462, LY3214996, and ulixertinib/BVD-523). Several inhibitors of each component of the PI 3-K-AKT-mTOR effector pathway have undergone development, with some receiving approval for the treatment of non-RAS-mutant cancers (the p110d inhibitor, idelalisib, and the mTORC1 inhibitors, temsirolimus and everolimus).72
As monotherapy, these inhibitors have not yet exhibited enough clinical advantage for RAS-mutant cancers. Preclinical mouse model research found that synchronous suppression of specific components of the PI 3-K and RAF effector pathways can engender powerful inhibition of tumor development. Nevertheless, these combinations have not demonstrated enough activity among clinical trials, with toxicity continuing to represent a restricting factor.
Additional validated RAS effectors comprise two that engender activation of further RAS superfamily proteins.1 RAS activation of RACGEFS or RALGEFs engenders activation of the RAC or RAL small GTPases, respectively. Small molecule inhibitors of RAC and RAL have been delineated. These small GTPases also use protein kinase effectors, PAK serine/threonine kinases for RAC and TBK1 for RalB, and inhibitors of these kinases have also been delineated.36,73
In recent years, rigosertib, initially developed as a non-ATP competitive multi-kinase inhibitor, has been classified as an inhibitor of effector binding to RAS, via its binding to the RA or RBD domains located on most RAS effectors.74 However, an additional study discovered an indirect mechanism with which rigosertib engendered activation of the JNK MAPK and indirect inactivation of ERK signaling.75 To summarize, although arguably the most encouraging direction for the development of clinically effectual anti-RAS therapies in the short term, effector inhibitors have displayed limited to no antitumor effectiveness as monotherapy. Rather, combination inhibitor strategies are likely required to overcome normal cell toxicity and the fast onset of resistance mechanisms.
Targeting Synthetic Lethal Interactors of Mutant RAS
One appealing approach for the development of RAS inhibitors has been the determination of synthetic lethal interactors embodying mutant but not wild-type RAS.76 This strategy is founded on the notion that a gene, the function of which is essential for cell viability only in the context of RAS mutations, offers a target for selective inhibition of RAS-mutant cancer cells. Earlier research determined protein kinases (e.g., STK33, TBK1) to be RAS synthetic lethal interactors.77–79 Due to the fact that protein kinases are more druggable than RAS itself, these studies generated substantial enthusiasm.
Unfortunately, succeeding research discovered that the proteins identified in many of these examinations were not robustly connected to mutant KRAS function, rendering them less attractive targets for selective targeting of RAS-mutant cancers. Despite these setbacks, there remains a strong belief that bona fide synthetic lethal interactors of mutant RAS embody strong potential as targets that can provide pharmacologic inhibitors selective for RAS-mutant cancers.
To overcome some of the limits of earlier research, enhanced cell models of KRAS dependency and experimental strategies are required.80 Many of the earlier studies used so-called “isogenic” matched pairs of human tumor cell lines. Established utilizing the heterozygous KRAS-mutant DLD-1 and HCT 116 CRC cell lines, matching lines were produced by stable deletion of the endogenous KRAS allele.
However, since the consequent lines lacking the mutant KRAS allele were clonally derived from a heterogeneous cell population, and because cells missing mutant KRAS have been adapted to overcome their addiction to mutant KRAS, these lines are not authentically isogenic. The screening of large panels of KRAS mutant and WT cell lines, to compensate for cell line heterogeneity, might overcome the limits of isogenic pairs. Synthetic lethal screens have been typically undertaken under anchorage-dependent culture conditions. Screens utilizing three-dimensional cultures under anchorage-independent conditions might detect genes with increased physiologic value. Finally, the important off-target activities of siRNA-based libraries are being dealt with by employing CRISPR/Cas9-based library screens.81 To summarize, although pursuing synthetic lethal interactors of mutant KRAS has not satisfied the field’s high expectations, it is possible that enhanced experimental models and strategies will overcome historic restrictions.
Exploiting RAS-Driven Metabolism
Cancer cells display enhanced energy and biosynthetic requirements to increase their heightened proliferative state. Therefore, cells reprogram metabolic processes to either repurpose and recycle intracellular fuel sources or scavenge extracellular components. Mutant RAS is directly involved in regulating specific cellular signaling cascades to achieve a shift towards anabolic metabolic processes producing nucleic acids, amino acids, lipids, and cofactors comprising NADPH for redox balance and reductive biosynthesis.8,82,83 As a consequence, elucidating the mechanisms by which RAS triggers altered metabolism might help illuminate therapeutic strategies for RAS-mutant cancers.
The discovery that cancerous tissue displays changed metabolism is credited to Otto Warburg, who in the 1920s observed that tumor tissues metabolize glucose to lactate at a rate that is ten times faster than normal cells.84 While the exact benefit the Warburg Effect has on cancer cells is still debated,85 aberrant metabolisms is now considered an indicator of cancer.86 Although RAS had been involved in the upregulation of glycolysis,87 the mechanistic foundation for this observation has only recently begun to be described (Figure 2).
Firstly, RAS was demonstrated to drive augmented glucose uptake by upregulating the GLUT1 gene encoding the glucose transporter, GLUT1.88 Moreover, in an inducible Kras mouse model of PDAC, Kras activation was linked to augmented Glut1 gene expression. Secondly, Kras also upregulates glycolytic enzymes (Hk1, Hk2, Ldha, and Pfkl), engendering increased conversion of pyruvate to lactate.25 Thirdly, RAS modulates genes encoding enzymes that route glucose intermediates into the hexosamine biosynthetic pathway (HBP; Gfpt1), thus supporting protein glycosylation, and the non-oxidative pentose phosphate pathway (PPP; Rpia, Rpe), thus supporting nucleotide biosynthesis.25 KRAS modulates these enzymes via signaling through the RAF-MEK-ERK pathway and eventually via regulation of MYC expression. In this way, inhibitors of the ERK MAPK pathway can be utilized to inhibit KRASdriven glycolytic perturbations.
The final stage of glycolysis is the efflux of lactate into the extracellular space. Since high intracellular lactate negatively affects further glycolytic activity, targeting lactate efflux is a possible therapeutic strategy. In fact, the inhibition of LDHA with the small molecule polyphenolic naphthalene (FX-11) augmented oxidative stress in LDHA-driven cancer cells, inhibiting the malignant advancement of human pancreatic xenografts in turn.89 Notably, additional research has suggested that effectiveness depends on TP53 mutation status, as mice bearing WT TP53 patient-derived tumors exhibited resistance to FX-11.90 Similarly, heterogeneity in response was observed with GNE-140, a pharmacologically distinct LDHA inhibitor.
However, variability in response was correlated to the tumor’s capacity to use oxidative phosphorylation (OXPHOS), suggesting that combining inhibitors of OXPHOS and glycolysis might increase clinical effectiveness.91 Metformin, a biguanide utilized in the treatment of type 2 diabetes, blocks hepatic glucose use. Although early retrospective analyses of cancer incidence in the diabetic population attracted considerable enthusiasm in regard to the prospect that metformin therapy reduced pancreatic cancer frequency, a significant amount of contradictory research has been published over the last half-decade.92
Moreover, two recent phase 2 clinical trials have observed no benefit of metformin when it is administered at the levels required for glycemic control.93,94 Trials are still ongoing to establish whether metformin can be utilized as maintenance therapy among patients with stable disease (NCT02048384) or if more powerful biguanides are more effective (NCT02475499). Additionally, Linus Pauling posited high-dose vitamin C as a cancer therapy in the early 1970s.95 At the time, this proposal received ridicule and seemed to have been proven wrong by two clinical trials in the 1970s and 1980s, which showed that orally administered high-dose vitamin C made no impact on patient survival.96,97
Nevertheless, recent evidence demonstrated that BRAF- and KRAS-mutant colorectal cancer cells not only exhibited enhanced uptake of the oxidized form of vitamin C (dehydroascorbate), but also that this engenders cell death as a result of oxidative stress.98 Moreover, this study showed that the methodology of drug delivery might have provided the basis for historic clinical failure, as intravenous administration was needed to attain the plasma concentrations required for a therapeutic effect. Additional research is needed to establish whether high-dose vitamin C is indeed a practical strategy for patients. RAS-mutant PDAC is also characterized by enhanced dependency on glutamine.99 While it has been proposed that glutamine intermediate metabolism in the mitochondria fuels processes external to bulk protein synthesis,100 contemporary macromolecular fractionation research has demonstrated that protein represents the primary endpoint for glutamine carbon.101
In the mitochondria, glutamine can be processed into glutamate via either transaminases or glutamate dehydrogenase 1 (GLUD1). In KRAS-mutant PDAC, glutamine-derived aspartate is carried to the cytosol where it undergoes glutamic-oxaloacetate transaminase-1 (GOT1)-dependent conversion to oxaloacetate, which eventually undergoes conversion to pyruvate.102 The byproduct of these reactions is an augmented NADPH/NADP+ ratio in the cell, which is critical for PDAC cells displaying downregulated oxidative pentose phosphate pathway activity. While a small molecule directly targeting this GOT1-dependent pathway is yet to undergo development, the glutaminase inhibitors Compound 968103 and bis-2-(5-phenylacetamido1,2,3-thaidiazol-2-yl) ethyl sulfide 3 (BPTES)104 lowered the proliferation of PDAC in vitro. Whenever treatment with BPTES or Compound 968 was combined with hydrogen peroxide treatment (to augment ROS), there was an observable increased effect.102
This shows that combining agents targeting glutamine utilization with ROS-generating treatments, including radiation, might represent a practical therapy strategy. Moreover, in combination with drugs inducing ROS formation, CB-839, which is an orally available GLS1 inhibitor, has been proven to synergistically shrink PDAC xenografts and augment overall survival of tumor-bearing mice.105 At present, CB-839 is being investigated in clinical trials for various cancer types.
A further option of glutamine entry into cancer cells is macropinocytosis, an endocytic scavenging mechanism by which cells ingest albumin and additional extracellular nutrients.106 Mutant RAS can drive increases in macropinocytosis,107, and human PDAC cell lines, in addition to a KRAS-driven mouse model of PDAC, exhibited enhanced macropinocytotic activity.108 Moreover, when labeled albumin is directly delivered to tissues and tumors in live mice, it is possible to directly observe macropinocytosis at higher levels in tumor tissue in comparison to the surrounding normal tissue.109
The internalized protein undergoes lysosomal degradation, generating essential amino acids, particularly glutamine, which subsequently enters the central carbon pool.108,110 While the increased macropinocytotic activity in PDAC is RAS-dependent, the processes driving this activity continue to be inadequately understood. The observation that mTORC1 signaling inhibits macropinocytosis is particularly notable.110 This finding might partially account for the insignificant impact of mTOR inhibitors on PDAC. This is because, in the context of these highly macropinocytotic cells, mTOR inhibition might drive further increased macropinocytosis.
In contrast to alternative endocytic mechanisms, macropinocytosis is suppressed by treatment with 5-[N-ethyl-N-isopropyl] amiloride (EIPA). EIPA, an inhibitor of Na+/H+ exchange, suppressed PDAC tumor formation in vivo.108 However, this tool compound cannot be utilized in humans, and there are still no specific inhibitors of macropinocytosis for cancer therapy in the clinic.
Nevertheless, there is a temptation to hypothesize that the success of nab-paclitaxel, an albumin-bound form of paclitaxel, in the therapy of PDAC is a partial consequence of the exploitation of high macropinocytotic levels among PDAC tumors.111,112 Moreover, as a result of the hypovascular nature of pancreatic tumors, PDAC cells are constantly nutrient-deprived, and they consequently possess evolved mechanisms of recycling intracellular nutrients.
Macroautophagy hereafter referred to as autophagy, is one metabolic process linked to RAS-mutant cancers. Among healthy cells, autophagy is a catabolic process of “self-eating”, with which surplus or dysfunctional cellular components, which include cellular organelles, undergo degradation. Extant tumors hijack autophagy as a means of supporting survival via the provision of amino acids, lipids, and nucleic acids.113 This scavenging mechanism is complex and requires the production of autophagosomes, and subsequent fusion with lysosomes, which enables their degradation and the release of nutrients. Autophagy is raised in PDAC.113,114
To support the cooperative role between autophagy and RAS, immortal, non-tumorigenic baby mouse kidney epithelial (iBMK) cells ectopically expressing oncogenic KRAS or HRAS were examined. In comparison to their non-transformed equivalents, these cells manifested enhanced defects in mitochondrial respiration following autophagy inhibition.114 Nonetheless, a cooperative relationship between autophagy and constitutive RAS activation is not exclusively observed. Expression of oncogenic HRAS in human fibroblast and ovarian epithelial cells engenders senescence and autophagy-mediated cell death, respectively, for example.115,116 In addition, the role of TP53 mutation status in regulating autophagy is not adequately understood.
In a PDAC mouse model harboring a Tp53 homozygous deletion, autophagy inhibited tumor progression.117 However, in a further mouse model driven by Tp53 loss of heterozygosity, which is closer to TP53 mutation among humans, Tp53 status did not demonstrably affect the protumorigenic role played by autophagy.118 Moreover, as the understanding of cellular crosstalk and intratumoral heterogeneity increases, there is emerging data to suggest that autophagy might play a role in the support provided by pancreatic stellate cells (PSCs) to their tumor cell neighbors. A contemporary study showed that autophagy was needed for the release of alanine from PSCs and that this alanine, taken up by PDAC cells, went beyond glucose and glutamine-derived carbon in respect of incorporation into mitochondrial metabolic processes.119
Treatment with chloroquine, which indirectly blocks autophagy through the inhibition of lysosomal acidification, weakens PDAC growth in vitro and in vivo. 113,118 Hydroxychloroquine (HCQ) has received approval by the FDA for the treatment of malaria and multiple rheumatological disorders for many years, and, consequently, clinical trials testing anticancer effectiveness proceeded rapidly. However, HCQ as monotherapy has been characterized by a general lack of success in the clinic, most likely as a result of the restricted power of HCQ to inhibit autophagy in vivo.120 Nevertheless, when HCQ was combined with gemcitabine, the PDAC patients that exhibited enhanced disease-free and overall survival were identified as those which were responsive to HCQ, as manifested by changes in LC3, a key component of the autophagy pathway and an acknowledged biomarker for HCQ responsiveness.121
The amalgamation of HCQ with additional chemotherapeutics is the foundation for several present clinical trials. HCQ suppresses the process of lysosomal acidification, which is not specific to autophagy and, moreover, inhibits additional cellular processes, including macropinocytosis. Therefore, research focus has also been directed towards earlier steps in the autophagy pathway which are kinase-regulated, and which might yield further targets for small molecule inhibitors.
The first such small molecule recorded, Spautin-1 (specific and potent autophagy inhibitor-1), promotes the degradation of VPS34 PI3- kinase complexes via inhibition of two ubiquitin-specific peptidases, USP10 and 13, and therefore engenders cancer cell death in the event of nutrient deprivation.122 However, VPS34 is critical for sorting multiple endocytic pathways and consequently, like HCQ, might not represent a specific target for autophagy inhibition. Therefore, attention has been directed towards ULK1 and 2 which function further upstream and might be more selective targets. An ULK1-specific inhibitor, SBI-0206965, has been demonstrated to specifically inhibit ULK-1 mediated phosphorylation events and to undergo synergization with mTOR inhibitors to kill tumor cells.123 In addition, an ULK1/2 inhibitor, MRT 68921, has been delineated and demonstrated to inhibit autophagy in cells.124 However, additional chemical optimization of each of these more specific autophagy inhibitors is required to render these compounds clinically satisfactory.
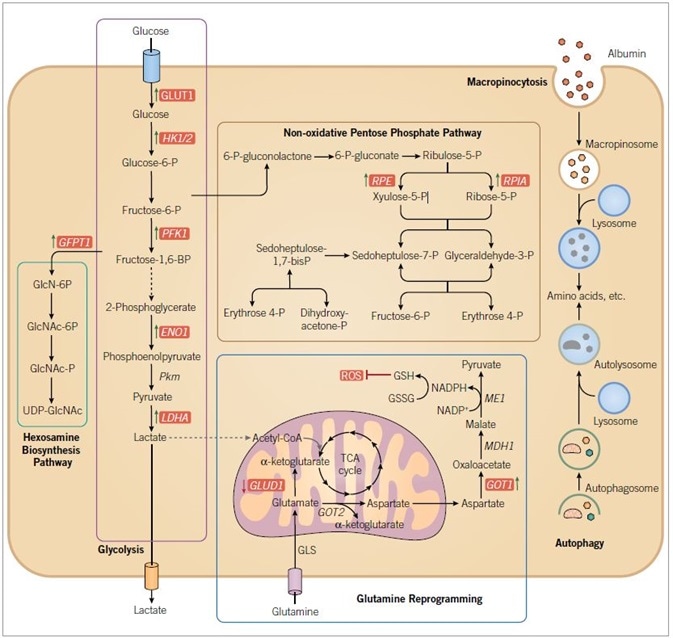
Figure 2. RAS-driven alterations in metabolism. RAS mutant cancer cells are characterized by increased macropinocytosis and uptake of albumin, leading to lysosomal degradation and the release of amino acids. RAS mutant cancer cells also exhibit altered autophagy, leading to degradation of organelles and proteins, and the production of amino acids and other components to support metabolism. Oncogenic KRAS directs glucose metabolism into biosynthetic pathways in PDAC by upregulating many key enzymes in glycolysis. Oncogenic KRAS induces nonoxidative PPP flux to fuel increased nucleic acid biosynthesis and activates the hexosamine biosynthesis and glycosylation pathways. PDAC cells also utilize a non-canonical pathway to process glutamine, through which it maintains redox balance and supports cell growth. Blue text indicates RAS-dependent gene and/or protein expression, with arrows indicating increased (green) or decreased (red) expression. Enzymes are indicated in italics. Abbreviations used are: GLUT1, glucose transporter 1; HK 1/2, hexokinase 1/2; PFK1, phosphofructokinase 1; ENO1, enolase 1; PKM, pyruvate kinase; LDHA, lactate dehydrogenase A; GFPT1, glucosamine-fructose-6-phosphate aminotransferase-1; GlcN, glucosamine; GlcNAc, N-acetylglucosamine; PPP, pentose phosphate pathway; RPE, ribulose-5-phosphate-3-epimerase; RPIA, ribulose-5-phosphate isomerase; GLUD1, glutamate dehydrogenase 1; GOT, aspartate transaminase; GLS, glutaminase; GSH, glutathione; GSSG, glutathione disulfide; ME1, malic enzyme; ROS, reactive oxygen species.
Future Prospects
More than 30 years of failures to generate effectual anti-RAS therapies is attributable to a general underestimation of cancer genetics and cancer heterogeneity, of the complexity of RAS function, of the dynamic nature of signal transduction networks. Cancer genome sequencing endeavors have shown that RAS continues to be the critical oncogene driver for cancers that represent the top three causes of cancer deaths in the US, and that collectively cause nearly 42% of deaths caused by cancer. There is a near-universal occurrence of RAS mutations in the most fatal of the fatal cancers, pancreatic cancer. Earlier failures engendered the erroneous notion that RAS is not druggable.
Therefore, although the successful clinical development of an effectual anti-RAS treatment continues to pose significant challenges, historic pessimism has become contemporary enthusiasm and vigor. What are the chances of success? Is the contemporary understanding of RAS adequately developed to avoid the problems of the past? As Yogi Berra asserted, “It’s tough to make predictions, especially about the future”. There is unlikely to be one simple anti-RAS therapy that will be effectual in all RAS-mutant cancers. The notable intra- and inter-patient heterogeneity within one cancer, and the cell context divergences seen among various RAS-mutant cancers, will necessitate more than one anti-RAS strategy, and these might eventually be further augmented by concurrent immunotherapy, an emergent area with regards to RAS.
Of the extant RAS-centric strategies, targeting effector signaling arguably embodies the strongest prospects for success over the succeeding five years. In terms of the ideal strategy, the development of direct RAS inhibitors, the route to transitioning the existing tool compounds to clinically effective drugs possessing adequate selectivity and potency might be a long one. Alongside enhanced technologies and models for screening, there is reserved enthusiasm that the once encouraging chances of mining for synthetic lethal interactors of mutant RAS will eventually accomplish their elevated promises to determine RAS mutant-selective treatments.
To summarize, there is now realistic enthusiasm that effectual anti-RAS therapies will, at last, be developed, although the type of therapies, as well as when they will be achieved, is less than certain. What is clear is that an improved awareness of RAS modulation and activity, and how these are integrated with additional physiological and pathophysiological functions, will be required to successfully detect and target RAS therapeutic vulnerabilities.
References and Further Reading
- Cox et al. (2014) Nat Rev Drug Discov 13 828.
- Stephen et al. (2014) Cancer Cell 25 272.
- Cox et al. (2010) Small GTPases 1 2.
- Rauen. (2013) Annu Rev Genomics Hum Genet 14 355.
- Wennerberg et al. (2005) J Cell Sci 118 843.
- Siegel et al. (2016) CA Cancer J Clin 66 7.
- Ledford. (2015) Nature 520 278.
- Bryant et al. (2014) Trends Biochem Sci 39 91.
- Fearon. (2011) Annu Rev Pathol 6 479.
- Ryan et al. (2014) N Engl J Med 371 2140.
- Hingorani et al. (2003) Cancer Cell 4 437.
- Aguirre et al. (2003) Genes Dev 17 3112.
- Hingorani et al. (2005) Cancer Cell 7 469.
- Kojima et al. (2007) Cancer Res 67 8121.
- Jones et al. (2008) Science 321 1801.
- Biankin et al. (2012) Nature 491 399.
- Sausen et al. (2015) Nat Commun 6 7686.
- Waddell et al. (2015) Nature 518 495.
- Witkiewicz et al. (2015) Nat Commun 6 6744.
- Bailey et al. (2016) Nature 531 47.
- Brummelkamp et al. (2002) Cancer Cell 2 243.
- Lim et al. (2008) Nature 452 646.
- Collins et al. (2012) J Clin Invest 122 639.
- Collins et al. (2012) PLoS One 7 e49707.
- Ying et al. (2012) Cell 149 656.
- Kapoor et al. (2014) Cell 158 185.
- Shao et al. (2014) Cell 158 171.
- Vetter. in Ras superfamily small G proteins: biology and Mechanisms 1 Vol. 1 (ed A. Wittinghofer) Ch. 2, 25 (Spinger-Verlag Wien, 2014).
- Cox et al. (2015) Clin Cancer Res 21 1819.
- Prior et al. (2012) Cancer Res 72 2457.
- Bos et al. (2007) Cell 129 865.
- Vigil et al. (2010) Nat Rev Cancer 10 842.
- Ryan et al. (2015) Trends Cancer 1 183.
- Fritsch et al. (2013) Cell 154 940.
- Bodemann et al. (2008) Nat Rev Cancer 8 133.
- Gentry et al. (2014) Biochim Biophys Acta 1843 2976.
- Lim et al. (2006) Curr Biol 16 2385.
- Halaban. (2015) Clin Ther 37 682.
- Heid et al. (2011) Gastroenterology 141 719.
- Kissil et al. (2007) Cancer Res 67 8089.
- Ahearn et al. (2012) Nat Rev Mol Cell Biol 13 39.
- Tsai et al. (2015) Proc Natl Acad Sci U S A 112 779.
- Bivona et al. (2006) Mol Cell 21 481.
- Sung et al. (2013) Proc Natl Acad Sci U S A 110 20593.
- Marcus et al. (2015) Clin Cancer Res 21 1810.
- Ostrem et al. (2016) Nat Rev Drug Discov 15 771.
- Welsch et al. (2017) Cell 168 878.
- Ostrem et al. (2013) Nature 503 548.
- Lim et al. (2014) Angew Chem Int Ed Engl 53 199.
- Lito et al. (2016) Science 351 604.
- Patricelli et al. (2016) Cancer Discov 6 316.
- Waldmann et al. (2004) Angew Chem Int Ed Engl 43 454.
- Muller et al. (2004) Angew Chem Int Ed Engl 43 450.
- Karaguni et al. (2002) Cancer Res 62 1718.
- Herrmann et al. (1998) Oncogene 17 1769.
- Pan et al. (2008) Cell Signal 20 1134.
- Berndt et al. (2011) Nat Rev Cancer 11 775.
- Marom et al. (1995) J Biol Chem 270 22263.
- Blum et al. (2008) Recent Pat Anticancer Drug Discov 3 31.
- Papke et al. (2016) Nat Commun 7 11360.
- Zimmermann et al. (2013) Nature 497 638.
- Martin-Gago et al. (2017) Angew Chem Int Ed Engl 56 2423.
- Cho et al. (2015) Mol Cell Biol 36 363.
- van der Hoeven et al. (2013) Mol Cell Biol 33 237.
- Foster et al. (2016) Cancer Cell 29 477.
- Chen et al. (2016) Cancer Discov 6 300.
- Collisson et al. (2012) Cancer Discov 2 685.
- Samatar et al. (2014) Nat Rev Drug Discov 13 928.
- Peng et al. (2014) Cancer Res 74 Abstract nr DDT02.
- Zhang et al. (2015) Nature 526 583.
- Morris et al. (2013) Cancer Discov 3 742.
- Fruman et al. (2014) Nat Rev Drug Discov 13 140.
- Baker et al. (2014) Clin Cancer Res 20 4740.
- Athuluri-Divakar et al. (2016) Cell 165 643.
- Ritt et al. (2016) Mol Cell 64 875.
- Downward. (2015) Clin Cancer Res 21 1802.
- Barbie et al. (2009) Nature 462 108.
- Luo et al. (2009) Cell 137 835.
- Scholl et al. (2009) Cell 137 821.
- Yu et al. (2013) Enzymes 34 Pt. B 201.
- Wang et al. (2017) Cell.
- White. (2013) Genes Dev 27 2065.
- Kimmelman. (2015) Clin Cancer Res 21 1828.
- Warburg. (1956) Science 123 309.
- Liberti et al. (2016) Trends Biochem Sci 41 211.
- Hanahan et al. (2011) Cell 144 646.
- Racker et al. (1985) Proc Natl Acad Sci U S A 82 3535.
- Yun et al. (2009) Science 325 1555.
- Le et al. (2010) Proc Natl Acad Sci U S A 107 2037.
- Rajeshkumar et al. (2015) Cancer Res 75 3355.
- Boudreau et al. (2016) Nat Chem Biol 12 779.
- Amin et al. (2016) Gut Liver 10 665.
- Reni et al. (2016) Clin Cancer Res 22 1076.
- Kordes et al. (2015) Lancet Oncol 16 839.
- Cameron et al. (1978) Proc Natl Acad Sci U S A 75 4538.
- Moertel et al. (1985) N Engl J Med 312 137.
- Creagan et al. (1979) N Engl J Med 301 687.
- Yun et al. (2015) Science 350 1391.
- Wise et al. (2010) Trends Biochem Sci 35 427.
- Daye et al. (2012) Semin Cell Dev Biol 23 362.
- Hosios et al. (2016) Dev Cell 36 540.
- Son et al. (2013) Nature 496 101.
- Wang et al. (2010) Cancer Cell 18 207.
- Robinson et al. (2007) Biochem J 406 407.
- Chakrabarti et al. (2015) Cancer Metab 3 12.
- Ha et al. (2016) Front Physiol 7 381.
- Bar-Sagi et al. (1986) Science 233 1061.
- Commisso et al. (2013) Nature 497 633.
- Davidson et al. (2017) Nat Med 23 235.
- Palm et al. (2015) Cell 162 259.
- Von Hoff et al. (2013) N Engl J Med 369 1691.
- Von Hoff et al. (2011) J Clin Oncol 29 4548.
- Yang et al. (2011) Genes Dev 25 717.
- Guo et al. (2011) Genes Dev 25 460.
- Elgendy et al. (2011) Mol Cell 42 23.
- Young et al. (2009) Genes Dev 23 798.
- Rosenfeldt et al. (2013) Nature 504 296.
- Yang et al. (2014) Cancer Discov 4 905.
- Sousa et al. (2016) Nature 536 479.
- Wolpin et al. (2014) Oncologist 19 637.
- Boone et al. (2015) Ann Surg Oncol 22 4402.
- Liu et al. (2011) Cell 147 223.
- Egan et al. (2015) Mol Cell 59 285.
- Petherick et al. (2015) J Biol Chem 290 11376.
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.