The P2 receptors for extracellular nucleotides participate in the regulation of almost every physiological process and are widely distributed in the body.1,2 Nucleotide receptors in the muscular, immune, cardiovascular, inflammatory, and central and peripheral nervous systems are of particular interest.
The ubiquitous signaling characteristics of extracellular nucleotides acting at two distinct families of P2 receptors – fast P2X ion channels and P2Y receptors (G-protein-coupled receptors) – are now well recognized. These extracellular nucleotides are generated as a result of cell damage and tissue stress and in the process of neurotransmitter release and channel formation. Their concentrations can differ massively depending on circumstances.
Consequently, the state of activation of these receptors can rely heavily on the disease states or stress conditions affecting a particular organ. The P2 receptors react to numerous extracellular mono- and dinucleotides (Table 1).
In agonist selectivity, the P2X receptors are more structurally restrictive than P2Y receptors. The P2Y receptors are activated by a group of five or more naturally occurring nucleotides, while the P2X receptors are activated principally by ATPs.
The P2X receptors are distributed throughout the nervous system (enteric, autonomic, and sensory neurons, cochlear, central, and retinal cells), vascular system (endothelium, cardiomyocytes, and smooth muscle), the pulmonary and digestive systems (epithelium and visceral smooth muscle), bone, skeletal muscle, and hematopoietic cells.
The P2X receptors are made up of trimeric ligand-gated ion channels. The subunits are numbered P2X1 to P2X7, and both homotrimers and heterotrimers occur. Activation of P2X receptors results in an influx of cations like calcium and sodium, which depolarize excitable cells and activate cytosolic enzymes respectively. Upon prolonged agonist exposure, the P2X7 receptor also opens a large pore, which can pass dye molecules and organic cations.
The knowledge of P2X receptor structures was recently advanced with the X-ray crystallographic determination of the P2X4 subunit.3,4 Yet, this structure did not determine the specific ligand binding site within the protein. A huge challenge in designing new agonist and antagonist ligands for a specific P2X receptor subtype is that the heterotrimers and homotrimers could possess totally different structural requirements.
The correspondence of the P2Y receptor subtypes along with their native nucleotide ligands can be seen in Table 1. Because the assignment of numbers to certain putative P2Y receptors was later shown to be premature, the numbering of unique human P2Y receptors has some gaps, with some of the sequences which were previously assigned being P2Y species homologs and others being other varieties of receptors.
Each of the native nucleotides can activate a variety of P2Y receptor subtypes. The structures of representative adenine (Figure 1A, 1–10) and uracil (Figure 1B, 11–28) nucleotides that activate P2 receptors are conveyed. The adenine nucleotide ATP is a full agonist at two human P2Y subtypes (P2Y2 and P2Y11 receptors), and the corresponding diphosphate ADP activates three different subtypes (P2Y1, P2Y12, and P2Y13 receptors).
The uracil nucleotide UTP activates two subtypes (P2Y2 and P2Y4 receptors), while UDP is now known to also activate P2Y14 receptors along with the originally designated native agonist UDP-glucose, it was previously thought to activate only a single subtype (P2Y6 receptors).5 Several P2 receptors are activated by the naturally occurring dinucleotide Ap4A and its homologs.
The structure, signaling, and regulation of P2Y receptors have been investigated, and subfamilies of P2Y1-like and P2Y12-like receptors have been realized. These subfamilies are made up of two pharmacologically distinct groups of P2Y receptors, which also correspond with similarities in the function of key amino acid residues.6
The preferential coupling of the first subfamily of P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11 receptors is to Gq, leading to the activation of phospholipase Cβ (PLCβ), and the second subfamily of P2Y12, P2Y13, and P2Y14 receptors couple to Gi leading to the inhibition of adenylyl cyclase. To stimulate adenylyl cyclase, P2Y11 receptors also activate Gs.
The comparisons of the structural characteristics and functionally important amino acid residues within the family have been outlined. Specific cationic residues and other residues in the TM region (for example Phe in TM3), and on the extracellular loops possess conserved functions within the P2Y family. Molecular recognition in the P2Y1, P2Y2, P2Y11, and P2Y12 receptors has been explored extensively via mutagenesis.7–10
P2Y receptor regulation has also been examined. The P2Y1 receptor is desensitized more quickly than the P2Y12 receptor in platelets, which express two ADP-responsive P2Y subtypes.11 Primarily, the P2Y1 receptor is desensitized through PKC-dependent processes, and the P2Y12 receptor is a good substrate for the GPCR kinases (GRKs) which result in arrestin binding.
Residues on the cytosolic C-terminal domain related to the regulation of the P2Y1 receptor have been inspected. The internalization of the P2Y11 receptor depends on coexpression of the quickly desensitizing P2Y1 receptor, proposing the occurrence of receptor dimers.12
A number of heterodimers of P2Y receptors with other P2Y and non P2Y GPCRs have been suggested. For example, a dimer of A1 adenosine receptors and P2Y1 receptors was characterized.13 Recurrent problems with the utilization of some P2 receptor ligands include cross-reactivity with numerous P2 receptors and low bioavailability because of polyanions, like sulfonates and phosphates which are present in the molecules.
Lability in biological systems is another issue of many of the currently used ligands. A big family of ectonucleotidase enzymes hydrolyzes the native nucleotides resulting in complications in the interpretation of biological results. In the final step by the action of CD73/5-nucleotidase on AMP, adenine nucleotides are progressively converted enzymatically, to create adenosine, which activates its own family of four receptors.
Selective inhibitors of ectonucleotidases that can act as modulators of receptor function are being investigated.14 Additionally, a lot of P2 receptor antagonists and agonists are known to inhibit ectonucleotidases at comparable concentrations. Known P2 antagonists frequently interact intracellularly with other signaling mediators, like G proteins.
Table 1. Subtypes of P2 receptors and their ligands (potency at the human homologs shown as pEC50, unless noted r = rat)
| Receptor |
Main Distribution |
Agonists (native in bold, pEC50) |
Antagonists |
| P2X1 |
Smooth muscle, platelets, cerebellum, dorsal horn spinal neurons |
BzATP 8.7 > ATP 7.3, 2-MeSATP 7.3, α,β-MeATP 6.7 (rapid desensitization) >> CTP 4.4 |
NF 449 9.5 > IP5I 8.8 > TNP-ATP 8.2 > Ro 0437626 > NF 279 7.7 |
| P2X2 |
Smooth muscle, brain, pancreas, retina, chromaffin cells, autonomic and sensory ganglia |
ATP 5.9, 2-MeSATP 5.8 ≥ 2-MeSATP 5.8 >> α,β-MeATP <4 |
RB2 6.4 (r), iso PPADS 6.4 (r) > PPADS 5.4 (r) > Suramin 4.5 (r) |
| P2X3 |
Nociceptive sensory neurons, NTS, some sympathetic neurons |
ATP 6.5, 2-MeSATP 6.5 ≥ Ap4A 6.3, α,β-MeATP 6.1 (rapid desensitization) |
TNP-ATP 9.0, iso PPADS 7.1 > A317491 7.6 > NF 110 7.4 > PPADS 5.8 |
| P2X4 |
Microglia, testis, colon, endothelial cells |
ATP 6.3 >> α,β-MeATP 5.1 >> CTP 3.5, Ivermectin (potentiates) 6.6 |
5-BDBD >> TNP-ATP 4.8, PPADS 4.6 > BBG 3.9 (r) |
| P2X5 |
Proliferating cells in skin, gut, bladder, thymus, spinal cord, heart, adrenal medulla |
ATPγS 6.2 (r), ATP 6.0 >> α,β-MeATP <5.2 |
BBG 6.3 > PPADS 5.6, Suramin 5.4 |
| P2X6 |
Brain, motor neurons in spinal cord |
(no functional homomultimer) |
– |
| P2X7 |
Macrophages, mast cells, microglia, pancreas, skin, endocrine organs |
BzATP 5.3 > ATP 4.0 ≥ 2-MeSATP >4 >> α,β-MeATP >4 |
KN 62 7.5, BBG 8.0 (r) |
| P2Y1 |
Brain, epithelial and endothelial cells, platelets, immune cells, osteoclasts |
MRS 2365 9.4 > 2-MeSADP 8.2 >> ADPβS 7.0 > ADP 5.1 > ATP |
MRS 2500 9.0 > MRS 2279 7.3 > MRS 2179 6.5 |
| P2Y2 |
Immune cells, epithelial and endothelial cells, kidney tubules, osteoblasts |
UTP 8.1, MRS 2698 8.1 ≥ ATP 7.1, INS 365 7.0 > INS 37217 6.7, UTPγS 6.6 > Ap4A 6.1 > MRS 2768 5.7 |
AR-C 126313 6 > Suramin 4.3 > RB2 >4 |
| P2Y4 |
Endothelial cells, placenta |
2′-azido-dUTP 7.1 > UTPγS 5.8, UTP 5.6 ≥ ATP 5.7 (rat), > Ap4A 5.5 > CTP 5.2, ITP 5.1 |
ATP (human) 4.4 > RB2 >4 > Suramin >4 |
| P2Y6 |
Airway and intestinal epithelial cells, spleen, placenta, T-cells, thymus |
MRS 2693 7.8 > UDPβS 7.3, PBS 0474 7.2 > INS 48823 6.9, Up3U 6.6, UDP 6.5 > UTP >> ATP |
MRS 2578 7.4 (noncompetitive) > RB2, PPADS |
| P2Y11 |
Spleen, intestine, granulocytes |
ATPγS 5.5 > ARC 67085MX 5.0 > BzATP 5.1 ≥ ATP 4.8 |
NF 157 7.4 > Suramin 4.8 > RB2 >4 |
| P2Y12 |
Platelets, brain (glial cells), microglial cells |
2-MeSADP 7.9 ≥ ADP 7.2 |
ARC 69931MX 8.4 > AZD 6140 7.9, INS 50589 7.8 > RB2 7.6 (r)> 2-MeSAMP 4.0 |
| P2Y13 |
Spleen, brain, lymph nodes, bone marrow |
ADP 7.9 = 2-MeSADP 7.9 > 2-MeSATP 7.1, ATP 6.6 |
ARC 69931MX 8.4 > ARC 67085 6.7 > MRS 2211 6.0 |
| P2Y14 |
Placenta, mast cells, adipose tissue, stomach, intestine, discrete brain regions |
MRS 2690 7.3 > UDP 6.8, UDPglucose 6.5 > UDP-galactose 6.2 |
– |
One of the causes of the relatively slow progress in identifying competitive antagonists of the P2 receptors is that there are not many selective radioligands available for either the P2Y or P2X receptors. Formerly, numerous radioactive nucleotides have been proposed to bind to certain P2 receptors, but a number of these reports were questioned later, and presently just the P2Y1, P2Y12, P2X1 and P2X3 receptors have viable radioligands.15–17
Hence, more versatile and improved affinity probes for the P2 receptors are still required. New selective antagonists and agonists have been identified recently for a few of the eight mammalian subtypes of P2Y receptors and for some of the seven mammalian subtypes of P2X receptors. Subtype-selective nucleotide agonists are a result of a cautious exploration of the structure-activity relationships (SARs) at relevant P2 receptors.
As a result of the conversion of agonists into antagonists, library screening, and the careful structural modification of known non-selective ligands, selective antagonist ligands for P2 receptors have been established. The structures of representative nucleotide (Figure 2A, 29–42) and non-nucleotide (Figure 2B, 43–73) antagonists of the P2 receptors are outlined.
Pharmacological Probes for P2X Receptors
The development of P2X receptor ligands for possible therapeutic applications has begun. Selective P2X receptor antagonists are of particular interest in diabetic retinopathy, urinary incontinence, pain control, inflammatory diseases such as rheumatoid arthritis, and some other conditions.
Non-Selective P2X Ligands
All subtypes of P2X receptors are activated by ATP, but at different concentrations varying from the low nanomolar through to the high micromolar.18 ADP and AMP, when purified, are inactive at P2X receptors. 2‑Methylthioadenosine 5′-triphosphate (2-MeSATP) is a potent agonist at multiple P2X receptors, for example, P2X1 (EC50 = 54 nM) and P2X3 (EC50 = 350 nM) receptors.
α,β‑Methyleneadenosine 5′-triphosphate (Figure 1A 6) activates and desensitizes the P2X1 receptor and is inactive at the P2X2 receptor. It acts as a radioligand of P2X1 and P2X3 receptors in tritiated form. Non-selective, older, and weak P2X antagonists (Figures 2A and 2B), for example, organic dyes 43 and 45, have been in use for decades.
Polysulfonated Suramin, the antiparasitic drug, and the pyridoxal phosphate derivatives PPADS and positional isomer isoPPADS are relatively nonsubtype-selective P2X antagonists, which also block several P2Y subtypes.19 At the P2X1 receptor, the PPADS analog MRS 2159 is more potent than PPADS and also antagonizes the P2X3 receptor. The nucleotide derivative TNP-ATP is a potent P2X antagonist which is selective for numerous subtypes.20
It antagonizes P2X1, P2X3 and heteromeric P2X2/3 receptors with respective IC50 values of 6, 0.9 and 7 nM, and exhibits 1000- fold selectivity for P2X3 over P2X2, P2X4 and P2X7 receptors. Evans Blue, the polysulfonated biphenyl derivative, behaves as a P2X receptor antagonist, but it also influences amino acid binding sites and other channels.
P2X1 and P2X2 Receptors
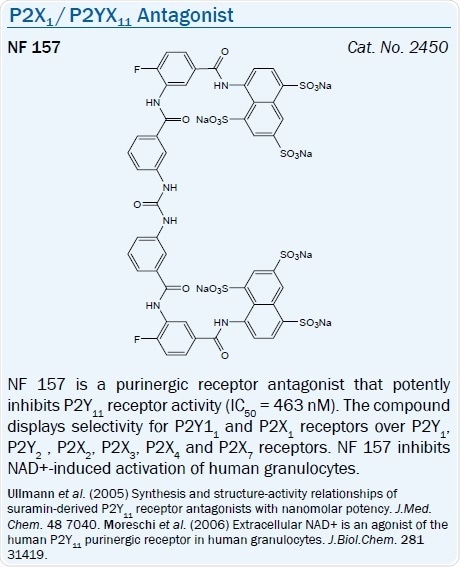
P2X1 antagonists have been seen in many compound classes. The Suramin derivative NF 157 for example, is a P2X1 antagonist which also inhibits the P2Y11 receptor.22
Some other Suramin derivatives which serve as selective P2X1 antagonists include: NF 279, PPNDS, and the more highly selective P2X1 antagonist NF 449.23,24 The earlier-reported Suramin analog NF 023 is a competitive, moderately selective, P2X antagonist with IC50 values of 0.21 and 28.9 μM at human P2X1 and P2X3 receptors respectively, and is inactive at P2X2 and P2X4 receptors.25
The benzimidazole-2-carboxamide derivative Ro 0437626 was recently said to be a selective P2X1 antagonist (IC50 = 3 μM) that exhibits > 30-fold selectivity over P2X2, P2X3 and P2X2/3 receptors (IC50 > 100 μM) in a separate chemical series.26 The dinucleotide Ip5I was seen to antagonize the P2Y1 receptor.42
A weak potentiator of P2X1-mediated responses is the pyridoxal phosphate derivative MRS 2219.27 There are no selective ligands for the P2X2 receptor. The nonselective antagonists RB-2, Suramin, isoPPADS, and TNP-ATP, have been utilized to examine this receptor.
P2X3 Receptor
In combination with P2X2 subunits, the P2X3 receptor can act as a heterotrimer or a homotrimer. The Suramin derivative NF 110 is a high-affinity P2X3 receptor antagonist (Ki values are 36, 82 and 4140 nM for P2X3, P2X1 and P2X2 receptors respectively) that is inactive at P2Y1, P2Y2 and P2Y11 receptors (IC50 > 10 μM). The introduction of the competitive antagonist by Abbott Laboratories was a notable advance.
A 317491, which blocks P2X2/3 (IC50 = 92 nM), and P2X3 (IC50 = 22 nM) receptors and, in comparison to P2X1 and P2X2 receptors, is roughly three orders of magnitude selective for P2X3.17 A 317491 is inactive at P2X4 receptors and at all P2Y receptors. A 317491 is of limited bioavailability because of the presence of three carboxylic acid groups.
The pyrimidinediamine derivative RO-3 is another potent P2X3 antagonist and is a selective antagonist of the heteromeric P2X2/3 and homomeric P2X3 receptors (pIC50 values are 5.9 and 7.0 nM respectively). It is inactive at P2X1, P2X2, P2X4, P2X5 and P2X7 receptors (IC50 > 10 M).29 The endogenous heptapeptide Spinorphin (LVVYPWT) was discovered to be an extremely potent P2X3 antagonist (IC50 = 8.3 pM).30
P2X4 Receptor
Not many of the known P2X antagonists act at the P2X4 receptor. The benzofurodiazepinone derivative 5-BDBD is an antagonist of P2X4-mediated currents (IC50 = 0.50 μM).31 Ivermectin, the bacteria-derived broad-spectrum antiparasitic agent, which is a macrocyclic lactone, is a positive allosteric modulator for the P2X4 receptor, but it influences some other ion channels too, for example, nicotinic acetylcholine receptors.32

Figure 1A. Adenine derivatives that have been useful as antagonists in the study of P2 receptors. (Bold text denotes compounds available from Tocris at time of publication)
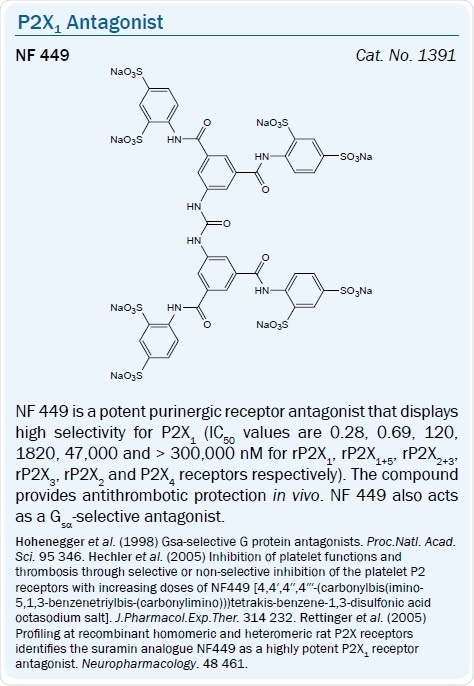
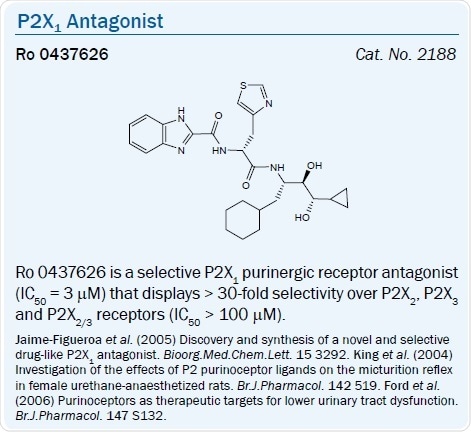
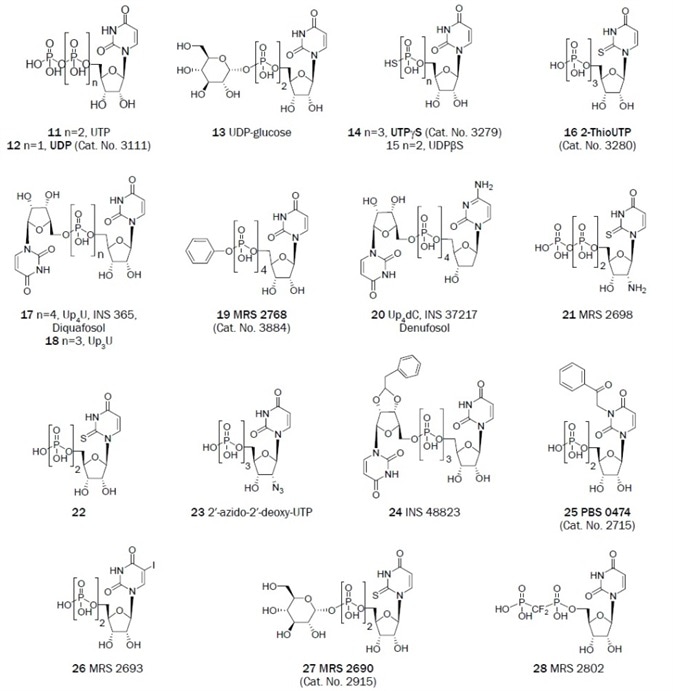
Figure 1B. Uracil derivatives that have been useful as antagonists in the study of P2 receptors. (Bold text denotes compounds available from Tocris at time of publication)
P2X5 Receptor
No selective ligands exist for the P2X5 and P2X6 receptors. Yet, the dye Coomassie Brilliant Blue G (BBG) has been effectively utilized to inhibit P2X5 receptor function (IC50 = 0.5 μM),33 but this dye also inhibits the P2X4 receptors (IC50 = 3 μM at human receptors) and P2X7 receptors (IC50 values are 10 nM and 267 nM at rat and human receptors respectively).
P2X7 Receptor
2′(3′)-O-(4-Benzoylbenzoyl)adenosine-5′-triphosphate (BzATP) is a P2X7 receptor agonist that displays an order of magnitude with higher potency than ATP. At P2X1 (pEC50 = 8.7) and P2Y1 receptors, it is also a partial agonist.34 The tyrosine and isoquinoline derivative KN-62 was one of the first antagonists of the P2X7 receptor identified but it acts in a non-competitive way and is inactive at the rat P2X7 homolog.
Additionally, KN-62 is an inhibitor of CaM kinase II.35 Oxidized-ATP (o-ATP) has also been extensively utilized to antagonize P2X7 receptors. It has been challenging to pinpoint antagonists which block the P2X7 receptor in a species-independent manner.
A selective, potent, and competitive P2X7 antagonist (pIC50 = 6.9) is the tetrazolylmethylpyridine derivative A 438079.36 The quinolinamino derivative A 740003 is a potent and selective competitive P2X7 receptor antagonist (IC50 values are 40 and 18 nM for human and rat P2X7 receptors respectively). In comparison to various P2X and P2Y receptors, that is also highly selective.36,37
Possessing KD values of 1.4 and 19 nM at human and rat P2X7 receptors respectively, the adamantyl derivative from AstraZeneca AZ 10606120 antagonizes the P2X7 receptor.38 The biphenyl derivative AZ 11645373 potently antagonized the human P2X7 receptor in a non-surmountable way with KB values between 5–20 nM and was inactive at the rat P2X7 receptor and at every other P2X subtype.39
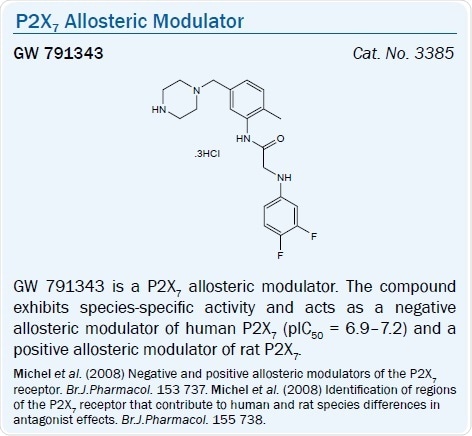
The substituted glycyl anilide derivative GW 791343 is a positive allosteric modulator of the rat P2X7 receptor and a negative allosteric modulator of the human P2X7 receptor (pIC50 = 6.9–7.2).40
Pharmacological Probes for P2Y Receptors
In the development of selective agonist and antagonist ligands for P2Y receptors for preclinical development progress has been made.2 Until now, the antithrombotic P2Y12 receptor antagonist Clopidogrel (Plavix) was the only P2Y receptor ligand in pharmaceutical use.41 Hence, there is a lot of activity to recognize new agents to act at the P2Y12 receptor and at other P2Y receptors for pharmaceutical development.
The fast-growing progress in this field has already lead to new drug candidates for dry eye disease, pulmonary diseases, and thrombosis. Numerous selective ligand probes, both antagonists and agonists of the P2Y receptors, are now available. Yet, some subtypes, for example, the P2Y4 receptor, are lacking such selective ligands entirely.
For P2Y1 and P2Y12 receptors, which both are proaggregatory in platelets, detailed SAR analyses have been created. Nucleotide agonists selective for P2Y1, P2Y2, P2Y6, and P2Y14 receptors and nucleotide antagonists selective for P2Y1 and P2Y12 receptors have been detailed. To avoid challenges of limited bioavailability and stability, selective non-nucleotide antagonists are now desired.
Such antagonists have been reported for P2Y1, P2Y2, P2Y6, P2Y11, P2Y12, and P2Y13 receptors. The screening of chemically diverse compound libraries has lead to competitive P2Y12 receptor antagonists which are being studied as possible antithrombotic agents.




P2Y1 Receptor
Similar to ADP, 2-MeSADP activates the P2Y1, P2Y12 and P2Y13 receptors. 2-MeSADP is a more potent agonist at the P2Y1 receptor than 2-MeSATP. Consistent with a small hydrophobic pocket in the P2Y1 receptor binding site surrounding the N6-position of adenine nucleotides, N6-methyl nucleotides are tolerated at the P2Y1 receptor.
The favored ribose-ring conformation for all of the subtypes of the P2Y1-like subfamily has been determined by utilizing conformationally-restricted (i.e. rigid) ribose equivalents, which also enhance the stability of the phosphate esters toward nucleotidases.
The methanocarba ring system made up of fused cyclopropane and cyclopentane has been particularly useful in looking at the biologically active conformations of nucleotide and nucleoside derivatives.
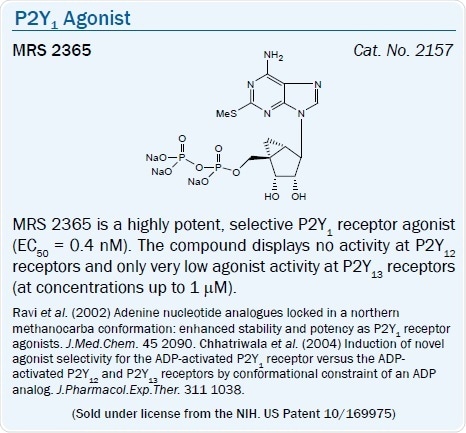
A high affinity, selective agonist of the P2Y1 receptor is the North (N)- methanocarba analog of 2-MeSADP, MRS 2365 (EC50 = 0.4 nM).11 The introduction of a borano group within the phosphate moiety of P2Y receptor agonists is a way of further enhancing hydrolytic stability.43
A number of nucleotide antagonists of the P2Y1 receptor have been introduced. Typically, these are adenine nucleotides that contain bisphosphate groups, for example, a ribose 3′,5′-bisphosphate moiety.
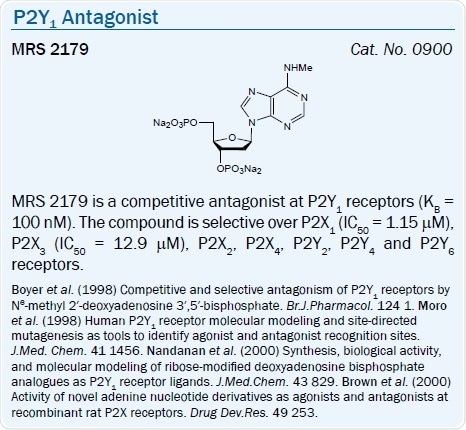
An N6‑methyl 2′-deoxyadenosine bisphosphate derivative MRS 2179 (pKB = 6.99) and its 2-chloro analog MRS 2216 are selective P2Y1 antagonists.44 The same (N)-conformational constraint of the ribose moiety which increases agonist action also favors the selectivity and potency in nucleotide antagonists.
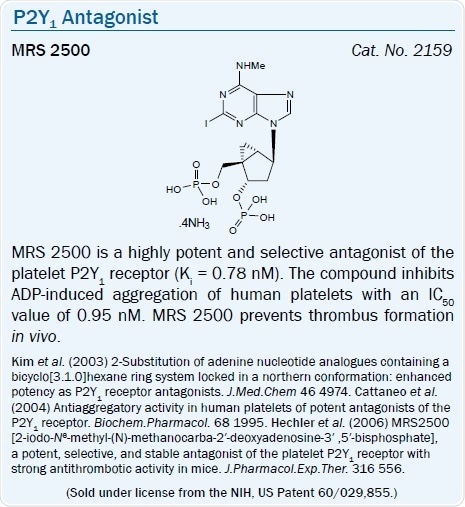
The ring-constrained (N)-methanocarba nucleotide bisphosphates MRS 2279 (pKB = 8.10) and MRS 2500 (pKB = 9) for example are high affinity, selective antagonists of the P2Y1 receptor.45 The antithrombotic action of MRS 2500 (by selectively blocking the P2Y1 receptor) has been shown in vivo in the mouse and also in some other species.46,47
Antagonists of the P2Y1 receptor of moderate affinity can also come from acyclic nucleotides, like the bisphosphates derivative MRS 2298.46 Non-nucleotide antagonists of the P2Y1 receptor have been found through the screening of structurally diverse chemical libraries.
The first of these compounds to be described was 63, which is an orally bioavailable and selective antagonist of the human P2Y1 receptor from Pfizer of novel chemotype with a Ki value of 90 nM.48 Some other structurally diverse antagonists of the P2Y1 receptor have also been revealed. Pyridyl isatogen (PIT) is an allosteric modulator of the P2Y1 receptor which exhibits mixed antagonism/potentiation.49
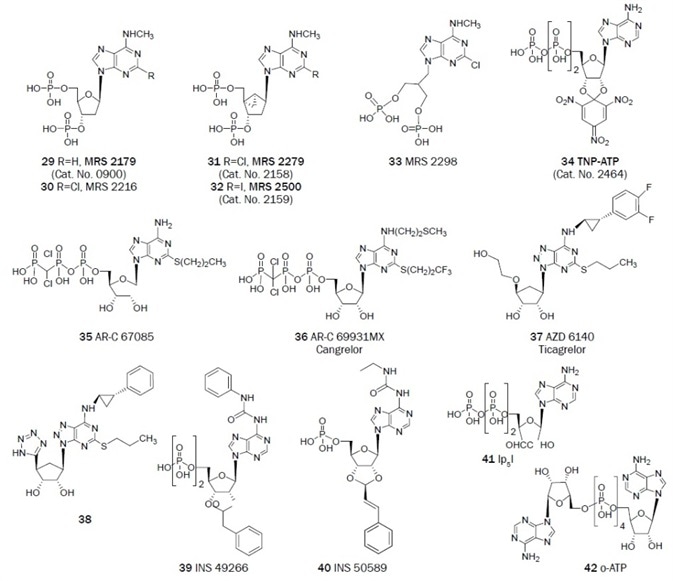
Figure 2A. Nucleotide derivatives useful as antagonists in the study of the P2 receptors. (Bold text denotes compounds available from Tocris at time of publication)
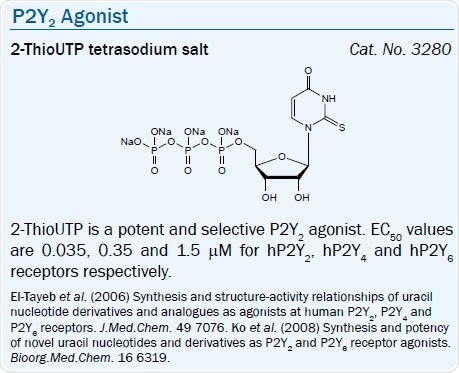
P2Y2 and P2Y4 Receptors
Both P2Y2 and P2Y4 receptors are activated by UTP, and simple modifications increase the selectivity for the P2Y2 receptor. A more stable and selective agonist of the P2Y2 receptor than UTP is UTPγS.50 Yet, it is subject to chemical oxidation. 2-ThioUTP is another selective agonist of the P2Y2 receptor.51
In comparison to the P2Y4 receptor, a combination of modifications of UTP in the selective P2Y2 agonist MRS 2698 supplied an EC50 of 8 nM and selectivity of 300-fold.52 Particular dinucleoside tetraphosphates activate the P2Y2 and P2Y4 receptors potently. The uracil dinucleotides which have been in clinical trials are Up4dC (INS 37217, Denufosol, EC50 = 0.22 μM) and Up4U (INS 365, Diquafosol, EC50 = 0.1 μM).53
Recently, in Japan, diquafosol was approved for use in treating dry eye. They are more stable to enzymatic hydrolysis than nucleoside triphosphates, by being dinucleotides, but compared to the P2Y4 receptor these agonists are non-selective. The 2′‑deoxycytidine (dC) moiety of 20 acts to increase the in vivo stability toward ectonucleotidases.
With moderate potency, the agonist MRS 2768 (uridine tetraphosphate δ-phenyl ester) is selective for the P2Y2 receptor (EC50 = 1.89 μM).54 Definitive antagonists of the P2Y2 receptor are unavailable. AR-C 126313 and its higher molecular weight analog AR-C 118925 were reported to selectively antagonize the P2Y2 receptor, yet it appears that these compounds are only micromolar in affinity (Figure 2B).55
The large polyanionic molecules Suramin and Reactive Blue 2 (RB2, an anthraquinone dye) are marginally selective antagonists of the P2Y2 and P2Y4 receptors, respectively. Yet, RB2 and Suramin also block numerous P2X receptors (Table 1). So, there are no truly selective ligands for the P2Y4 receptor.
5-fold P2Y4 selectivity was displayed by the agonist 2′-azido-2′-deoxy-UTP (Figure 1B 23).51 Hence, new antagonists and agonists are required to pharmacologically distinguish this subtype from the P2Y2 receptor, which is also activated by UTP. ATP, the other native agonist of the P2Y2 receptor, serves as an antagonist at the human, but not the rat, P2Y4 receptor.
P2Y6 Receptors
Both the P2Y6 and P2Y14 receptors are activated by UDP. However, it is worth noting that extracellular UDP can act as a substrate for the production of UTP through the action of nucleoside diphosphokinase (NDPK), which can complicate pharmacological examinations. The action of UPD at the P2Y14 receptor has been controversial.
Initially, UDP was detailed as inactive at the newly cloned P2Y14 receptor, but later study discovered an antagonist action of UDP at the human but not rat P2Y14 receptor. Lastly, the antagonist action of UDP took place in cells expressing an unnatural, engineered chimeric G protein. Yet, UDP acts as a potent agonist in HEK293 and other cells in which endogenous Gi proteins mediate the functional response.5
Hence, if multiple P2Y subtypes are present, inaccurate results could be gathered by utilizing UDP alone in pharmacological studies. UDPβS (15) and Up3U have been employed as more stable activators of the P2Y6 receptor subtype than UDP,50,53 although UDPβS activates the P2Y14 receptor as well.
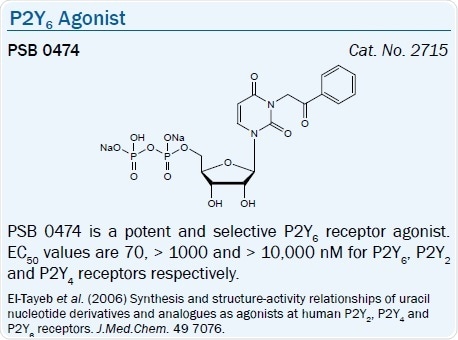
The SAR of nucleotide derivatives in activating the P2Y6 receptor has been examined. Specific dinucleoside triphosphates have been looked at as P2Y6 receptor ligands, for example, INS 48823 (EC50 = 125 nM) activates the receptor potently.56 Other UDP derivatives, e.g. 5-iodo-UDP (MRS 2693), and 3-phenacyl UDP (PSB 0474), are selective P2Y6 agonists with respective EC50 values of 15 and 70 nM.57,58
Molecular modeling estimated that the South (S)-conformation of the ribose ring is the preferred conformation in receptor binding, which by synthesis of a conformationally constrained methanocarba analog of UDP, was then confirmed. The noncompetitive P2Y6 receptor antagonist MRS 2578 is a diisothiocyanate derivative, which has low solubility and stability in liquid.59 Competitive antagonists of the P2Y6 receptor have not been reported as of yet.
P2Y11 Receptors
ATPγS (Figure 1A) serves as a potent P2Y11 receptor agonist. The P2Y12 antagonist 2-propylthio-β,γ-dichloromethylene-ATP (AR-C 67085, Figure 2A) is also the most potent agonist of the P2Y11 receptor (EC50 = 8.9 μM) reported.60 So, it must be employed cautiously in pharmacological studies in which both P2Y subtypes could be present.
Derived from nonselective P2 antagonist Suramin, a potent antagonist NF 157, has been said to be a selective antagonist of the P2Y11 receptor (pKi = 7.35).61 Yet, this compound also antagonizes the P2X1, P2X2, and P2X3 receptors.
P2Y12 Receptors
The medicinal chemistry of the P2Y12 receptor has been extensively explored. The thienopyridines, such as Clopidogrel (Figure 2B), act as liver-activated prodrugs that are irreversible inhibitors of the P2Y12 receptor.62 This thienopyridine P2Y12 receptor antagonist requires a two-step preactivation in vivo and therefore has a delayed onset of action and long reversal of the platelet effect after drug administration is stopped.
Prasugrel is another thienopyridine antagonist that has been present in clinical trials. It is a more potent P2Y12 antagonist but exhibits a longer bleeding time. Prasugrel only needs one step of preactivation in vivo.63 Directly-acting P2Y12 receptor antagonists have also been reported.
The introduction of numerous 5′-triphosphate analogs as selective receptor probes and clinical candidates has been enabled because of the observation that ATP serves as an antagonist at this ADP-activated subtype. So, AstraZeneca’s antithrombotic nucleotide derivatives from AR-C 67085 (EC50 = 30 μM) and ARC 69931MX (Cangrelor, EC50 = 0.4 nM) have been clinically tested as antithrombotic agents.64
A 5′-triphosphate group in adenine nucleotides is not a strict requirement for P2Y12 receptor antagonism, similar to the instance of compound 38 and the potent antagonist and approved medicine ticagrelor (AZD 6140 pIC50 = 7.9).64,65
Nucleotide derivatives from Inspire Pharmaceuticals, INS 49266 (an ADP derivative with EC50 of 52 nM) and INS 50589 (an AMP derivative with EC50 of 11 nM) are some other nucleotide antagonists of the P2Y12 receptor which have been discovered.66 The identification of novel chemotypes that act as P2Y12 receptor antagonists has been aided greatly by library screening, and many of these compounds are being developed by the pharmaceutical industry.
One very potent and selective competitive antagonist of the P2Y12 receptor, which was reported recently, is PSB 0739, derived from RB2.
P2Y13 Receptors
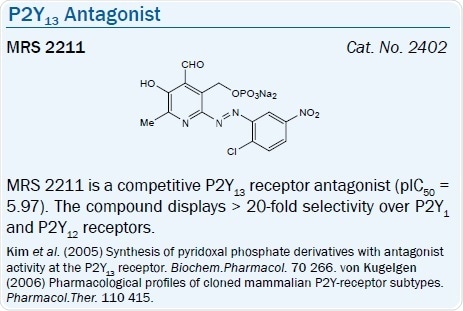
ADP is also the favored agonist ligand at the P2Y13 receptor, and ATP is less potent. ADP is 3-5-fold more potent than 2-MeSADP in the rat. A selective P2Y13 receptor antagonist, MRS 2211, a derivative of PPADS, has a pKi of 6.0 at this receptor.67
P2Y14 Receptors
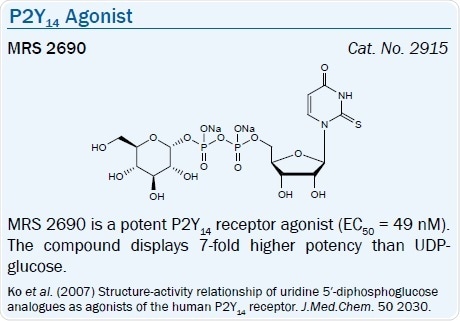
Recently, the SAR of analogs of UDP-glucose (EC50 = 0.35 μM) and UDP at the P2Y14 receptor was systematically examined.68 Other naturally occurring UDP-sugars activate this receptor less potently. MRS 2690, the 2-thio analog of UDP-glucose, is a 6-fold more potent agonist for the P2Y14 receptor and is inactive at the P2Y2 receptor, unlike UDP-glucose.
Concerning alteration of the ribose, nucleobase, and phosphate moieties of agonist ligands, the P2Y14 receptor is structurally restrictive. Yet, the glucose moiety may be deleted in UDP analogs, some of which are still extremely potent in receptor activation. For example, α,β‑difluoromethylene-UDP, MRS 2802, is inactive at the P2Y6 receptor and fully activates the human P2Y14 receptor with an EC50 of 63 nM.
Conclusion
Novel ligands for the P2Y and P2X receptor families are now available for employment as tools in pharmacological studies. Although generally of low bioavailability and stability in vivo, selective nucleotide agonist ligands, have been designed. Recently, as a result of library screening, conversion of agonists into antagonist, and the careful structural modification of known non-selective ligands, selective antagonist ligands for P2 receptors have been reported.
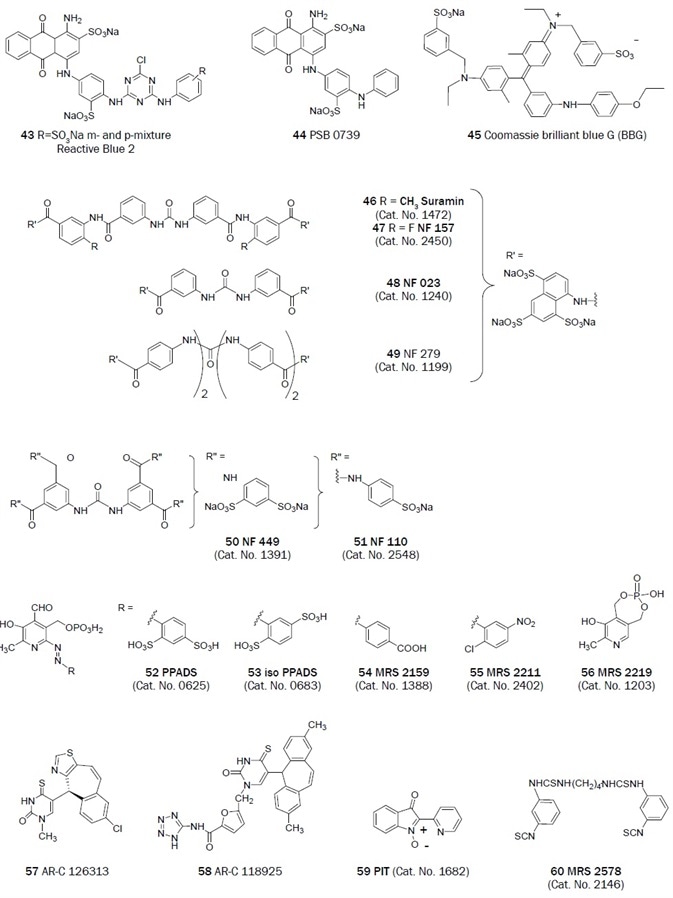
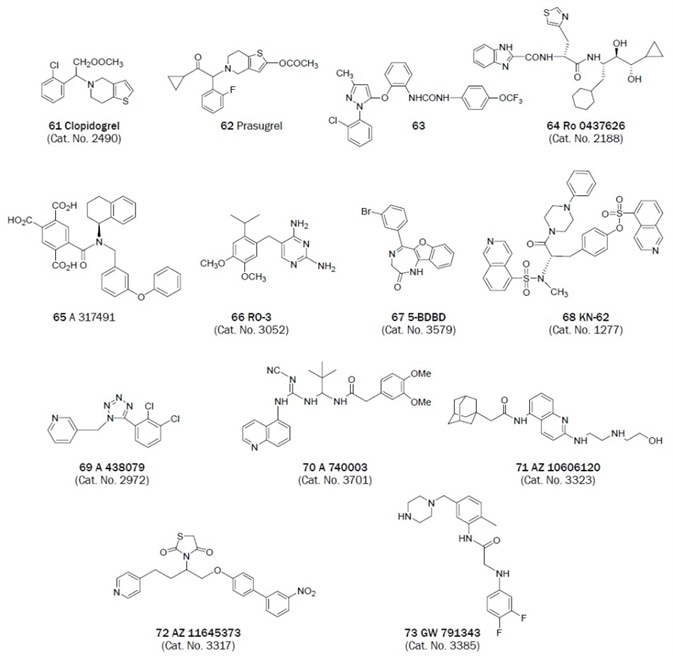
Figure 2B. Non-nucleotides that have been useful antagonists in the study of P2 receptor. (Bold text denotes compounds available from Tocris at time of publication)

Special Mentions
This white paper was based on the original work of:
Kenneth A. Jacobson: Kenneth Jacobson serves as Chief of the Laboratory of Bioorganic Chemistry and the Molecular Recognition Section at the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health in Bethesda, Maryland, USA. Dr. Jacobson is a medicinal chemist with interests in the structure and pharmacology of G-protein-coupled receptors, in particular receptors for adenosine and for purine and pyrimidine nucleotides.
Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, USA.
Email: [email protected]
References and Further Reading
- Surprenant and North (2009) Ann.Rev.Physiol. 71 333.
- Abbracchio et al. (2006) Pharmacol. Rev. 58 281.
- Kawate et al. (2009) Nature 460 592.
- Young (2009) Trends Biochem.Sci. 35 83.
- Carter et al. (2009) Mol.Pharmacol. 76 1341.
- Ivanov et al. (2006) J.Comput.Aided Mol.Des. 20 417.
- Hoffmann et al. (1999) J.Biol.Chem. 274 14639.
- Hillmann et al. (2009) J.Med.Chem. 52 2762.
- Zylberg et al. (2007) Biochem.J. 405 277.
- Hoffmann et al. (2009) J.Pharmacol.Exp.Ther. 331 648.
- Bourdon et al. (2006) J.Thromb.Haemost. 4 861.
- Ecke et al. (2008) Biochem.J. 409 107.
- Nakata et al. (2003) Drug Dev. Res. 58 340.
- Baqi et al. (2010) J.Med.Chem. 53 2076.
- Houston et al. (2006) Br.J.Pharmacol. 147 459.
- El-Tayeb et al. (2005) Bioorg.Med.Chem.Lett. 15 5450.
- Jarvis et al. (2004) J.Pharmacol.Exp.Ther. 310 407.
- Jacobson et al. (2002) J.Med.Chem. 45 4057.
- Kim et al. (2001) J.Med.Chem. 44 340.
- Virginio et al. (1998) Mol.Pharmacol. 53 969.
- Whittenburg et al. (1996) Naunyn-Schmiedebergs.Arch.Pharmacol. 354 491.
- Ullmann et al. (2005) J.Med.Chem. 48 7040.
- Rettinger et al. (2000) Neuropharmacology 39 2044.
- Rettinger et al. (2005) Neuropharmacology 48 461.
- Soto et al. (1999) Neuropharmacology 38 141.
- Jaime-Figueroa et al. (2005) Bioorg.Med.Chem.Lett. 15 3292.
- Jacobson et al. (1998) J.Med.Chem. 41 2201. 2
- Hausmann et al. (2006) Mol.Pharmacol. 69 2058.
- Carter et al. (2009) Bioorg.Med.Chem.Lett. 19 1628.
- Jung et al. (2007) J.Med.Chem. 50 4543.
- Donnelly-Roberts et al. (2008) J.Pharmacol.Exp.Ther. 324 409.
- Silberberg et al. (2007) Neuron 54 263.
- Bo et al. (2003) Mol.Pharmacol. 63 1407.
- Bianchi et al. (1999) Eur.J.Pharmacol. 376 127. References
- Chessell et al. (1998) Br.J.Pharmacol. 124 1314.
- Donnelly-Roberts et al. (2009) Br.J.Pharmacol. 157 1203.
- Michel et al. (2008) Br.J.Pharmacol. 153 737.
- Stokes et al. (2006) Br.J.Pharmacol. 149 880.
- Michel et al. (2008) Br.J.Pharmacol. 155 738.
- Cattaneo (2010) Circulation 121 171.
- King et al. (1999) Br.J.Pharmacol. 128 981.
- Eliahu et al. (2009) Eur.J.Med.Chem. 44 1525.
- Nandanan et al. (1999). J.Med.Chem. 42 1625.
- Kim et al. (2003) J.Med. Chem. 46 4974.
- Cattaneo et al. (2004) Biochem.Pharmacol. 68 1995.
- Hechler et al.. (2006) J.Pharm.Exp.Therap. 316 556.
- Pfefferkorn et al. (2008) Bioorg.Med.Chem.Lett 18 3338.
- Gao et al. (2004) Biochem.Pharmacol. 68 231.
- Hou et al. (2002) Am.J.Phys.Heart and Circ.Phys. 282 784.
- Jacobson et al. (2006) Biochem.Pharmacol. 71 540.
- Ivanov et al. (2007) J.Med.Chem. 50 1166.
- Shaver et al. (2005) Purinerg. Sig. 1 183.
- Ko et al. (2008) Bioorg.Med.Chem. 16 6319.
- Kindon et al. (1998) World Pat. 98 54180.
- Korcok et al. (2005) J.Biol.Chem. 280 16909.
- El-Tayeb et al. (2006) J.Med.Chem. 49 7076.
- Besada et al. (2006) J.Med.Chem. 49 5532-5543.
- Mamedova et al. (2004) Biochem. Pharmacol. 67 1763.
- Communi et al. (1999) Br.J.Pharmacol. 128 1199.
- Ullmann et al. (2005) J.Med.Chem. 48 7040.
- Savi et al. (2002) Throm.Haemostas. 84 891.
- Jakubowski et al. (2007) Br.J.Clin.Pharmacol. 63 421.
- Springthorpe et al. (2007) Bioorg.Med.Chem.Lett. 17 6013.
- Ye et al. (2008) ChemMedChem 3 732.
- Douglass et al. (2008) J.Med.Chem. 51 1007.
- Kim et al. (2005) Biochem.Pharmacol. 70 266.
- Das et al. (2010) J.Med.Chem. 53 471.
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.