During the in vitro culture of cells, small molecules are often used to manipulate signaling pathways. These signaling pathways are important targets for small molecules in the culture of stem cells that control cell proliferation and differentiation. It can be useful to target pathways such as the canonical Wnt, transforming growth factor-β (TGF-β) and retinoic acid signaling pathways to increase and maintain the proliferation of stem cells. Alternatively, this can be done to guide stem cell fate toward specific lineages in controlled differentiation.
This article offers a brief insight into the small molecules that interact with the primary signaling pathways which govern stem cell proliferation and differentiation to mediate stem cell behavior, as well as how small molecules play certain roles in the dedifferentiation of somatic cells to create populations of pluripotent stem cells.
The defining factor of stem cells is their ability to self-renew and their potential to differentiate into defined cellular subtypes.1 Four main types of stem cell exist; embryonic stem (ES) cells, induced pluripotent stem (iPS) cells, adult stem (AS) cells and cancer stem (CS) cells. Historically, ES cells derive from the inner cell mass of the developing blastocyst.
Upon exposure to developmental cues, ES cells can differentiate into any cell type, representing all three of the developing germ layers. Useful therapeutic tools and insight into key developmental processes may be provided by the study of ES cells.2 Through either forced expression of transcription factors, or exposure to a multitude of molecules that revert them back to a stem cell-like phenotype, iPS cells are derived from the reprogramming of somatic cells.3
iPS cells are thought to have a similar potency to ES cells, as they are both pluripotent and able to differentiate into cell types representing all three germ layers. This may provide therapeutic potential as treatment for degenerative diseases for autologous transplantation of iPS cell-derived cell types.
AS cells are conventionally responsible for the maintenance and repopulation of cell types found within specific niches in tissues, and have a much more limited differentiation potential. For instance, the hematopoietic stem cell found inside the bone marrow, that can give rise to only cell types found within the blood is an AS cell.4
The CS cell, the final category of stem cell, is responsible for the proliferation of cells in specific kinds of tumor. These cells may constitute a potential therapeutic target for anti-cancer drug development as they are thought to be implicated in cancer progression, initiation and metastasis.5 An integral part of stem cell research are the synthetic and naturally occurring molecules that interact with certain signaling pathways.
One can use compounds that are designed to interact with specific stages in developmental pathways to evoke a particular cellular response, which can then be modulated through compound concentration changes. The controlled differentiation of stem cells into specific cellular lineage can be achieved by the selectivity of molecules to act only upon the desired pathway. This is vital for in vitro modeling, which is used for basic research, drug screening and the study of pathological mechanisms. Two approaches which are frequently used to discover small molecules that interact with key signaling pathways are the custom design of molecular structures and screening of compound libraries.
Identification of Small Molecules
Several approaches can be used to identify small molecules that manipulate cell fate (reviewed by Lyssiotis et al).6 One of the most prevalent methods is high-throughput cell-based phenotypic screening, which involves the screening of large chemical libraries using immortalized cell lines.
A relatively straightforward example of high-throughput screening are reporter-based cellular assays: these involve the expression of a fluorescent reporter gene that is stimulated by the promoter of the gene of interest. Work by Kumagai et al constitutes an example of the use of reporter-based assays in stem cell research. They are screened for small molecules that promote human ES cell self-renewal to retain populations of undifferentiated cells.7
The key processes involved included observing green fluorescent protein expression driven by the Oct4 promoter, a transcription factor and marker of pluripotency, to determine how or if the small molecules tested affected cell fate.7 Though this method is largely well-suited to screening large libraries of molecules, the potential exists for the production of false positives and results still require rigorous testing.6
The multi-parametric, high-content image-based assay is another high-throughput screening method of small molecule identification, which involves the analysis of a desired phenotype at a single-cell level. This process is effective, but costly and time consuming.6
The size of libraries screened using high-throughput approaches can range from large collections of >2 million compounds (conventionally held by pharmaceutical companies) to smaller libraries of <10,000 compounds, (generally used in academic research and known to act on specific pathways). When screening with cells that have a limited viability, for instance primary cultures, smaller libraries are usually used, whereas larger libraries tend to be screened using immortalized cell lines that can easily be applied to a 384 or 1536 well format.6
A more profitable alternative to high-throughput screening is a more direct approach to the design of small molecule modulators. This would involve a more detailed study and analysis of the target with rational molecule design. To achieve this, analysis is undertaken of a small group of compounds with known biological activity to determine how they modulate specific pathways.
Microarray gene expression analysis, protein expression analysis and affinity-based target assays are all common assays used to elucidate these methods. Once known, the molecule’s mechanism can be related to the structure and subsequently used to design more effective compounds for more focused trials. EC 23, a synthetic retoid compound and potent inducer of stem cell differentiation, was developed as a result of structural design guided by biological activity.8
Pathways that Modulate Stem Cell Activity
It is important to modulate pathways controlling stem cell proliferation and differentiation to maintain a pool of self-renewing, undifferentiated cells, or to coax the fate of the stem cell down specifically selected lineages. Manipulation of many developmental pathways can take place to effect stem cell proliferation and differentiation, specifically those which feature in embryonic patterning, determination of cell fate and differentiation. It is vital to understand the molecular mechanisms of these processes, which can be attained either by designing molecules to act at specific stages in the pathway, or by screening a library of small molecules known to interact with the pathway.
It has been found that naturally occurring small molecules can interact with important developmental pathways. Modulation of the effects on cell fate can be achieved by changing the concentration of such molecules, enabling controlled differentiation. Development of multiple small synthetic molecules has taken place to act on primary developmental pathways, mediating stem cell behavior with great effect and increased potency. Below is an outline of a collection of the primary developmental pathways which are targeted by small molecules and involved in stem cell proliferation and differentiation.
Retinoic Acid Pathway
A major developmental pathway with significant roles in patterning and differentiation, especially in the developing nervous system, is the retinoic acid (RA) pathway (Figure 1).9 In the developing nervous system, RA is an important patterning factor, and acts in a concentration-dependent way in order to contribute toward anteroposterior and dorsoventral patterning of the neural plate and neural tube.9 Although RA is an important signaling molecule herein, RA also has significant parts to play in lung, pancreas and limb development, which illustrates the importance of this pathway in embryonic development.10
RA is a metabolite of vitamin A which is attained through dietary vegetables and meats, and circulated in the bloodstream as retinol bound to retinol-binding protein 4 (RBP4). Retinol is obtained by target cells through the membrane receptor’s (STRA6’s) action, which promotes the entry of retinol into the cytoplasm of the target cell.9
Retinol binds to retinol-binding protein once in the cytoplasm (RBP1), before it is metabolized to all-trans retinoic acid (ATRA). This is a two-step process which involves the activity of the two enzymes retinol dehydrogenase 10 (RDH10) and retinaldehyde dehydrogenases (RALDHs). Newly synthesized ATRA then binds to cellular retinoic-acid-binding protein 1 or 2 (CRABP1, CRABP2) and it can then be released from the cytoplasm to act on target cells in a paracrine or autocrine manner.9
CRABP2 assists the translocation of RA to the nucleus. Once inside the nucleus, RA binds to a transcription complex. This transcription complex includes a heterodimer of a retinoic acid receptor (RAR) and a retinoic X receptor (RXR).9 Various genes encode these RAR and RXR receptors, which include RARA, RARB and RARG, and RXRA, RXRB and RXRG respectively.
A sequence of DNA known as the retinoic acid response element (RARE) is bound by combinations of these receptors to induce transcription of target genes. Subsequently, after the activation of RARs/RXRs, RA leaves the nucleus and the CYP26 class of enzymes metabolizes it in the cytoplasm.9
To form neural subtypes, the role of RA in embryonic neural differentiation can be captured in vitro using the tool of ATRA with which to differentiate stem cells in culture. Tanoury et al provide an example of this, in their use of ATRA to induce neural differentiation of mouse ES cells, observed through expression of the pan-neuronal marker β-III-tubulin.
This permitted the researchers to study the molecular mechanism of neuronal differentiation.11 Despite this, there are limits on the use of naturally occurring ATRA in vitro thanks to the molecule’s instability and sensitivity to light and heat. These factors result in isomerization and breakdown of the molecule into other biologically active compounds.12
A problem is posed herein when using ATRA for in vitro differentiation studies, because changes in temperature and exposure to light are not always easy to avoid within a laboratory. In order to circumvent this variability, development of the synthetic retinoid EC 23 occurred as a stem cell differentiation tool, which is completely stable and mimics the biological activity of ATRA (Box 1).13 A potent inducer of stem cell differentiation, EC 23 regulates neural development14 and activates key proteins involved in the retinoic acid signaling pathway without degrading upon exposure to light.8
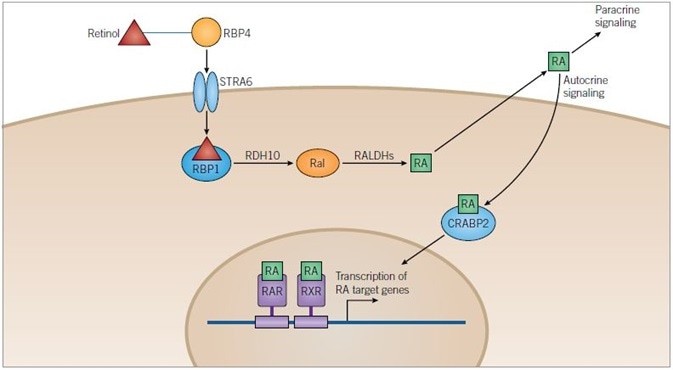
Figure 1. Retinoic Acid Pathway. Image credit: Tocris Bioscience
Retinoic Acid Pathway: Retinol is transported in the blood bound to retinol-binding protein 4 (RBP4) and enters cells through the transmembrane receptor STRA6. Once in the cytoplasm retinol binds to retinol-binding protein 1 (RBP1) and is metabolized to retinoic acid (RA) in a two-step process catalyzed by retinol dehydrogenase 10 (RDH10) and retinaldehyde dehydrogenases (RALDHs), with retinaldehyde (Ral) as an intermediate.
RA can be released from the cell and can either have a paracrine action on target cells or an autocrine action upon the cell that has metabolized it. RA is translocated into the nucleus of the target cell with the help of cellular retinoic-acid binding protein 2 (CRABP2) where it can bind retinoic acid receptors (RARs) and retinoid X receptors (RXRs) that can heterodimerize. Heterodimers of RXRs and RARs bind to DNA at a sequence known as the retinoic acid response element (RARE), activating the transcription of target genes.
Hedgehog Pathway
Many developmental processes are regulated by the Hedgehog (Hh) signaling pathway including neural cell fate and digit formation in a dose-dependent and tissue-specific manner (see review by Ingham and McMahon for details).15 Where a Hh ligand is absent, Patched (Ptch 1) catalytically inhibits the translocation of Smoothened (Smo) to the membrane,16 stopping it from inhibiting kinases such as protein kinase A (PKA), glycogen synthase kinase 3 b (GSK-3β) and casein kinase 1 (CK1).
The phosphorylation of the transcription factors Gli1/2/3 by PKA, GSK-3β and CK1 is thereby enabled, which results in proteasomal degradation of Gli2/3 to Gli2-R and Gli3-R, with Gli1 remaining at full length. Gli3-R is then translocated to the nucleus, wherein it inhibits the transcription of Hh target genes, while the inhibitory protein Suppressor-of-Fused (SuFu) sequesters remaining Gli,17 permitting only inactive Gli to travel to the nucleus.
Where they are present, Hh ligands bind to and inhibit Ptch1, permitting Smo to be translocated to the membrane where it inhibits the phosphorylation of Gli1/2/3. The result is that Gli1/2/3 is activated to Gli1/2/3-A, which then travels to the nucleus and stimulates the transcription of Hh target genes (Figure 2). Various divergent modifications of the Hh ligand activate different developmental pathways. This is the case, for instance, in mammals, where Sonic Hh regulates neural cell fate and Indian Hh regulates digit formation.
Many small molecules are available that modulate the Hh pathway, the majority of which act on Smo (Box 2). Cyclopamine, a naturally occurring plant-derived steroidal alkaloid extracted from corn lily, was the first discovered to affect the pathway, and directly inhibits Smo.18,19 Certain types of cancer have been linked to dysfunction of the Hh pathway. Therefore potential chemotherapeutic effects have been found in antagonists of Smo.
For instance, effectiveness has been demonstrated against basel cell carcinomas in phase 1 clinical trials by the small molecule GDC 0449.20 Through high-throughput screens of chemical libraries such as SANT1-, other antagonists of Smo have been discovered.22 High-throughput screening has also revealed Smo agonists, including purmorphamine, discovered during a screen of osteogenic compounds and proven to modulate various neural patterning.23,24
Identification of a family of Smo agonists (SAGs) which promote neuronal differentiation from mouse ES cells and which can induce growth of hair on mouse skin has also taken place.18 This is in addition to the discovery of small molecules that target the Hh pathway at alternative sites to Smo, including Robotnikinin, which is one of the only molecules to act upstream of Smo.25
Robotnikinin, a 12-membered macrocycle compound, was discovered during assays to identify recombinant Sonic Hh ligand binding molecules. It binds to the Hh ligand, which induces a conformational change that prevents binding to Ptch 1, this therefore inhibits the Hh pathway.26 Identification of small molecules that act downstream of Smo has also taken place. Gli antagonists that inhibit Gli-dependent transcription of Hh target genes include JK 184 and GANT.
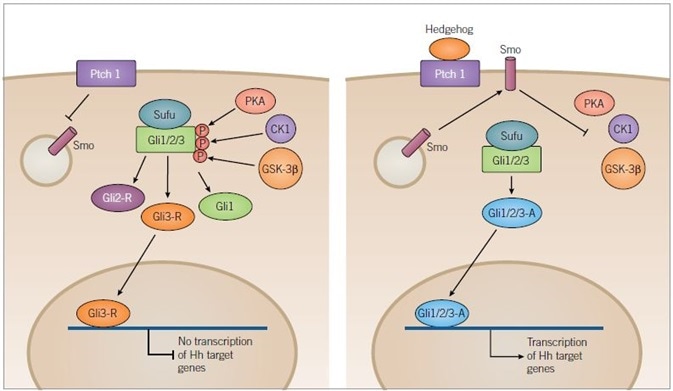
Figure 2. Hedgehog Signaling Pathway. Image credit: Tocris Bioscience
In the absence of ligand binding (left) Ptch1 inhibits translocation of Smoothened (Smo) to the membrane, resulting in the phosphorylation of the Sufu-Gli1/2/3 complex by the kinases, PKA, CK1 and GSK-3β. Phosphorylation results in the truncation of Gli1/2/3 forming inactive Gli2-R and Gli3-R, along with full-length Gli1.
Gli3-R is translocated to the nucleus and inhibits the transcription of target genes. In the presence of a Hedgehog (Hh) ligand (right) Ptch1 is inhibited and Smo is released to the membrane where it inhibits kinase activity. PKA, CK1 and GSK-3β are no longer able to phosphorylate Gli1/2/3 allowing it to remain in its full-length active form. Active Gli1/2/3 then travels to the nucleus where it can induce transcription of Hh target genes.
Transforming Growth Factor-β Superfamily
The TGF-β superfamily has over 30 ligands which consist of TGF-β, bone morphogenetic proteins (BMPs), nodal ligands, activins, and related proteins. Important developmental signaling cascades that regulate tissue differentiation and development through their roles in cell proliferation, differentiation and migration are activated by these ligands, as well as having significant tasks in adult homeostasis.28
Divergent ligand subsets have different developmental roles; for instance, nodal ligands are significant in embryogenesis and the formation of the mesoderm and endoderm germ layers, whereas BMPs have more to do with the differentiation of skin, neurons and bone.29,30 By binding of a ligand to the TGF-β family complex of receptors at the cell surface that are made up of two type I and two type II serine/threonine kinases, TGF-β signaling is initiated.
Ligand binding induces transphosphorylation of the type I receptor by the type II kinase which is activating it, and additionally promotes the phosphorylation of Smad2/3, which then complexes with Smad4.28 Translocation to the nucleus then takes place for the heterooligomeric complex of Smads, where it is able to regulate the transcription of TGF-β target genes through association with transcription factors and co-activators such as CBP or p300 (Figure 3). Smad6/7 can negatively regulate this pathway, which act as a negative feedback mechanism to prevent the phosphorylation of Smad2/3 and 4 by forming stable associations with activated type I receptors.31
To date, the majority of the small molecule inhibitors that target the TGF-β superfamily pathway that have been designed act on the type I kinase of the heterotrimeric receptor complex (Box 3). SB 431542 is the name of a small molecule which effects multiple biological processes including the following: proliferation, differentiation, and promotion of sheet formation of endothelial cells derived from ES cells.32
This is attained through its blocking of the action of activin receptor-like kinases 4, 5 and 7 (ALK4, TGF-βR1 and ALK7 respectively), which are type I receptor serine/threonine kinases (other small molecules including A 83-01 and SB 505124 do the same).
Differentiation of glioblastoma CS cells33 has been a proven effect of SB 431542, as well as its replacement of one of the factors used to generate iPS cells.34 This means that it is a significant tool in myriad aspects of stem cell research. Contrastingly, for most TGF-β superfamily modulators, the small molecule ITD 1 acts through promoting the proteasomal degradation of the type II receptor. It selectively targets TGF-β signaling instead of any of the other members of the TGF-β superfamily. In vitro, ITD 1 has been used to promote the differentiation of cardiomyocytes from ES cells.35
Dorsomorphin is the name given to an antagonist of the pathway, which is identified by a whole organism zebrafish developmental screen.36 The compound enhances myocardial differentiation from mouse ES cells, and blocks ALK 2, 3 and 6 which are associated with the BMP pathway. IDE 1 and 2 are the names given to two alkyl hydrazone derivatives that are agonists of TGF-β signaling, which can, through activation of TGF-β signaling, induce differentiation of endoderm from ES cells.37
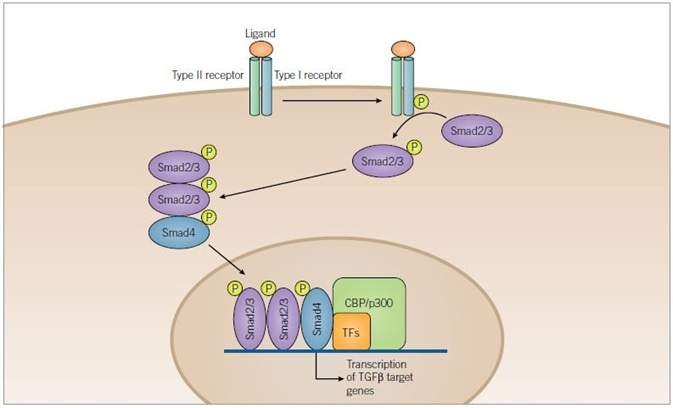
Figure 3. TGF-β Signaling Pathway. Image credit: Tocris Bioscience
TGF-β superfamily ligands bind to a serine/threonine kinase receptor complex of type I and type II subunits. Transphosphorylation of the type I receptor by the type II receptor is achieved through activation of the receptor complex. Before being translocated to the nucleus, Smad2/3 is then phosphorylated by the activated type I receptor and complexes with Smad4. Once inside the nucleus, the activated Smad complexes regulate transcription of target genes through interaction with transcription factors (TFs) and co-activators such as CBP or p300.
Canonical Wnt Pathway
One of the most studied biological signaling pathways is the canonical Wnt pathway. This pathway has major roles in proliferation, self-renewal and the differentiation of stem and progenitor cells during development.38 The pathway has been shown to have a central role in bone formation, hematopoiesis and neural differentiation. However, the risk is that dysregulation of the pathway can potentially invite the onset of cancer, thanks to the increased activation leading to increased cellular proliferation.
Various types of cancer, including breast, brain and colon cancers, have implicated the Wnt pathway and it has been suggested the pathway plays a role in their malignancy. The most prevalent genetic alteration in colorectal carcinomas are mutations to molecules central to the pathway (reviewed by Reya and Clevers).38
The central involvement of the canonical Wnt pathway in both stem cell biology and cancer pathogenesis makes it a desirable target for small molecule modulation. The binding of Wnt ligand stimulates signaling of the canonical Wnt pathway (Figure 4) to a receptor complex which incorporates a member of the Frizzled family and a member of the LDL-receptor family (Lrp5/6).38
A destruction complex forms due to the association of the scaffolding proteins adenomatous polyposis coli (APC) and axin when Wnt receptors are unoccupied, and the kinases CK1α and GSK-3β. The main cytoplasmic signaling molecule in the pathway is β-catenin, is bound to axin and sequentially phosphorylated by CK1α and GSK-3β, targeting it for proteasomal degradation.38
T-cell factor/lymphoid enhancer factor (Tcf/Lef) in the nucleus associates with co-repressors such as Groucho to inhibit the transcription of Wnt target genes, thus decreasing stem cell renewal and proliferation in the absence of Wnt. The actions of the destruction complex is inhibited through a Disheveled (DVL) dependent mechanism, which results in a cytoplasmic accumulation of β-catenin in the presence of Wnt ligand. β-catenin is then translocated to the nucleus where it can engage Tcf/Lef, along with docking proteins such as Legless and co-activators from the PYGO family, transiently converting Tcf/Lef into transcriptional activators.
The Tcf/Lef-β catenin-Legless-PYGO is the effector of the canonical Wnt pathway, which is responsible for promoting the transcription of target genes.39 Identification of several small molecules that can inhibit or activate Wnt signaling (Box 4), with targets for modulation including CK1α, GSK-3β and β-catenin itself has taken place thanks to screening of chemical libraries has identified several small molecules.
To antagonize the pathway, the benzothiazole derived compounds have been synthesized and split into two separate groups relating to their mechanism: IWP compounds that inhibit Wnt processing and secretion through inactivation of Porcupine (PORCN), and IWR compounds that promote the breakdown of β-catenin by enhancing the activity of the destruction complex.40
Axin, the concentration-limiting factor of the destruction complex, is stabilized by IWR compounds. A tankyrase inhibitor, XAV 939, antagonizes the pathway in a similar way.41 It has been proven to stunt the growth of APC-deficient colorectal cancer cells, highlighting the compound’s therapeutic potential and underlining the significance of understanding and targeting such pathways. Another small molecule, calphostin C (PKF 115584), was identified in such a screen, and proven to target the interaction of β-catenin with Tcf/Lef to inhibit Wnt signaling.42
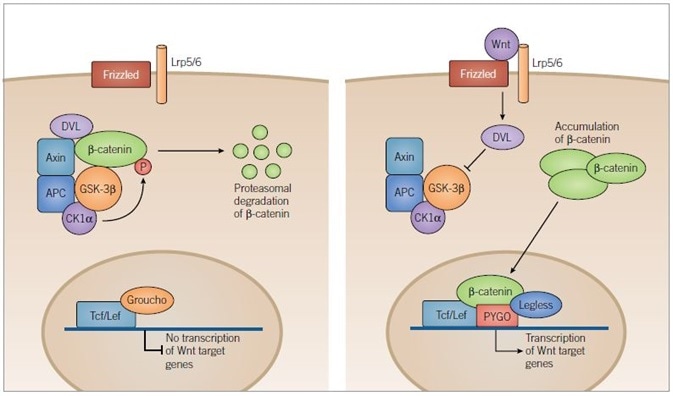
Figure 4. Canonical Wnt Pathway. Image credit: Tocris Bioscience
In the absence of Wnt signaling (left) adenomatous polyposis coli (APC) and axin complex and bind newly synthesized β-catenin, forming a destruction complex. Two kinases within the destruction complex, CK1α and GSK-3β, phosphorylate β-catenin targeting it for proteasomal degradation. In the nucleus, T-cell factor/lymphoid enhancer factor (Tcf/Lef) DNA-binding proteins repress target gene transcription through association with repressor proteins such as Groucho.
In the presence of Wnt ligand (right) Frizzled and Lrp5/6 are activated and the destruction complex is inhibited through a Dishevelled (DVL) dependent mechanism, preventing the phosphorylation of β-catenin. β-catenin then accumulates in the cytoplasm and is translocated to the nucleus where it engages transcription factors, Tcf/Lef and docking proteins of the Legless family, while associating with members of the PYGO family of co-activators. The β‑catenin-Tcf/Lef-Legless-PYGO nuclear complex promotes transcription of Wnt target genes.
The most common class of compounds that activate the Wnt pathway are the GSK-3β inhibitors, such as CHIR 99021.43 The deactivation of the destruction complex achieves this, therefore promoting β-catenin accumulation within the cytoplasm. Given that upregulation of the kinase is associated with the pathogenesis of multiple cancers as well as diseases such as type II diabetes and Alzheimer’s disease, GSK-3β is a desirable drug target. Considering this fact, a chemical library of over 4,000 compounds were screened. This was to identify the novel compound 3F8, which, through inhibition of GSK-3β, promotes Wnt signaling, with greater potency than other commonly used GSK-3β inhibitors.44
Similarly, the inhibition of CK1α can be achieved by compounds such as D 4476 and (R)-CR8 to prevent degradation of β-catenin and promote Wnt signaling. Through the action of the small molecule, prostaglandin E2 that acts via cAMP/PKC activity, β-catenin too can also be stabilized.45
Fibroblast Growth Factor and Notch Signaling Pathways
In a similar manner to the TGF-β superfamily, fibroblast growth factors (FGFs) are a large family of secreted molecules which enjoy myriad biological roles in embryonic development and the homeostasis of adult cells.46 By the complex expression of FGF ligands, various signaling pathways can be activated. Such pathways are tightly regulated, thanks to the fact that cancer can form when dysfunction arises.47 In vitro in the culture of ES cells, FGF ligands are frequently used, either to maintain cells in an undifferentiated state48 or to promote neural differentiation of cultures.
FGF signal transduction commences with the binding of ligands to their specific receptors, which results in dimerization and phosphorylation of multiple tyrosine residues on the receptors through the intrinsic tyrosine kinase activity of the receptors themselves. Receptor activation leads to the recruitment of signaling complexes which themselves activate a process of phosphorylation events.49
The specific ligand and receptor involved determine the specific molecules involved in this signaling cascade; however, three main pathways are involved in FGF signaling. These include: phospholipase C γ (PLCγ), phosphoinositide-3-kinase (PI 3-K) and mitogen-activated protein kinase (MAPK) pathways. Small molecules usually target the initial stages of the pathway (Box 5), thanks to the diversity of FGF signaling and its interaction with other signaling pathways. Various small molecules have been designed to inhibit the FGF receptor, including PD 173074,50 FIIN 151 and PD 161570.52
Specific pathways within FGF signaling constitute other targets for modulation; for instance, the MAPK pathway can be inhibited by the small molecule PD 0325901, which has undergone phase 2 clinical trials for use as a chemotherapeutic agent against certain types of cancer53, and which may also be used in the formation of iPS cells.54
The creation of specific cellular niches requires the Notch signaling pathway, thanks to the ability of its ligands to only activate signaling in adjoining cells instead of the surrounding area. The eural cell niche is an example of this, and possesses a balance between differentiated neural cells and progenitor cells.55 To develop small molecules that modulate this pathway has proven complex, in large part because many of the molecular targets are not unique to the Notch pathway. Nevertheless, DAPT56 and MRK 00357 are two antagonists of Notch which have been shown to reduce neural differentiation and promote programmed cell death respectively.
Promotion of ES Cell Self-Renewal
A problem is posed by spontaneous differentiation, in the long-term culture of ES cells in vitro, which requires the complicated mix of proteins and exogenous factors in culture media to inhibit differentiation through the promotion of pathways (such as the MAPK pathway). Typically, human ES cells require the addition of basic FGF into the medium, as well as being cultured on a feeder layer of mouse embryonic fibroblasts (MEFs) or in conditioned media to promote their continued self-renewal.
The use of human ES cells in regenerative medicine may be restricted by the use of MEFs as a feeder layer, due to concern of xenogenic contamination. Such contamination would likely risk consistency of results, as well as restricting the use of ES cells on a large scale. Though the exact identities of the active factors within the feeder layer remain unknown, work is being done to develop protocols to promote the proliferation of ES cells without the need for feeder layers.58
In a similar vein, mouse ES cells require the addition of leukemia inhibitory factor (LIF) and BMP 4 to their culture media: this addition may vary results due to the use of different batches. Stem cell fate may also be biased towards specific lineage types due to the use of serum products and feeder layers, through the activation of certain signaling pathways (reviewed by Xu et al)59. This factor has caused many groups to turn to high-throughput screening of small molecules, to overcome these issues (Box 6).
CHIR 99021, a GSK-3β inhibitor that suppresses the Wnt pathway, and PD 0325901 which inhibits the MEK pathway, are just two compounds which have been found to promote self-renewal in mouse ES cells. Pluripotin, a dihydropyrimidine, promotes the long-term maintenance of mouse ES cells without requiring feeder layers, LIF, BMPs or Wnt proteins.60
Pluripotin acts in a manner unrelated to the main stem cell signaling pathways, including the Wnt and BMP pathways. More structure-function investigations revealed that pluripotin binds to Ras GTPase activating protein (RasGAP) as well as extracellular signal-related kinase 1 (ERK1), two proteins with differentiation-inducing activity. The fact that Pluripotin simultaneously inhibits RasGAP and ERK1 and suppresses their roles in differentiation is enough for the long-term self-renewal of mouse ES cells.
Similarly, ID 8 is an indole derivative proven to promote mouse ES cell proliferation in serum-free conditions.61 6-bromoindirubin-3′-oxime (BIO), a potent inhibitor of GSK-3β, has been found to promote self-renewal in human ES cells via modulation of the Wnt pathway.62
However, it has been revealed that TWS 119, another GSK-3β inhibitor, induces neural differentiation of ES cells,63 which portrays the complexity of these signaling pathways. IQ 1 is another modulator of the Wnt pathway that has functions in the long-term maintenance of ES cells: it has been shown to encourage long term expansion of ES cells and prevent spontaneous differentiation.64 The development of small molecules such as these is deeply significant in that it can maintain undifferentiated ES cells and promote their proliferation. This, in turn, reduces variability of culture conditions and eliminates animal products from culture media to advance research into the therapeutic benefit of stem cell therapy.
Somatic Cell Reprogramming
In 2006, Yamanaka’s group reprogrammed mouse somatic cells into pluripotent populations of cells, that were able to differentiate into cells representative of the three embryonic germ layers.65 This somatic cell reprogramming results in a similar phenotype to that of ES cells.
Each of the pluripotent populations formed have a similar capacity for differentiation and are termed iPS cells. These iPS cells are considered to have potential therapeutic benefits. This is because they could be derived from a patient’s own somatic cells, reprogrammed and subsequently differentiated into specific cellular subtypes, to use in regenerative therapies and autologous transplantation.
In vitro disease modeling could also be impacted greatly by this avenue of stem cell research. In theory, somatic cells could be retrieved from an individual with a specific disease, and these cells reprogrammed and differentiated, with the resulting cells used in determining mechanisms of pathogenesis.
Certain ethical concerns of nuclear transfer and the use of ES cells could also be met and addressed with this development of iPS cells. Nonetheless, before the further development of iPS cell-based therapies, optimization of the reprogramming protocol must take place as the yield of iPS cells obtained is usually small, and the timeframe is weeks to generate populations of cells.
Traditionally, reprogramming protocols incorporate exogenous genetic manipulation of key pluripotency transcription factors such as Oct4, Sox2, Klf4 and c‑Myc using retroviruses. This is termed the four-factor method. Despite this, to consider iPS cell-based therapies for use in medicine, we must eliminate the use of retroviruses and transcription factors associated with tumorigenesis. This goes some way to explaining why, rather than genetic manipulation through transcription factors, chemically defined approaches to reprogramming somatic cells have been developed.
Screens of small molecules that have the potential to replace one or all these transcription factors have been conducted to achieve this (Box 7). One of these screens analyzed the ability of compounds to stimulate dedifferentiation of lineage-committed myoblasts; dedifferentiated cells were subsequently stimulated to differentiate into specific cellular subtypes. Such a screen identified Reversine, a small molecule that is a 2,6‑disubstituted purine analog.66
Reversine was shown to act via MEK signaling and non-muscle myosin II heavy chain, and proven to have dedifferentiation activity on cell types (including dermal fibroblasts with high efficiency in vitro and in vivo).67 The application of small molecules that target histone modifications has improved iPS cell generation, like valproic acid and trichostatin A, both of which inhibit histone deacetylases (HDACs).
The yield of iPS cells has been enhanced by Valproic acid and trichostatin A through the traditional four-factor dedifferentiation method; however, valproic acid has also been shown to enhance three-factor reprogramming (minus c-Myc)68 and two-factor reprogramming (without c-Myc and Klf4).69 Also, DNA and histone methylation constitute other targets for small molecules in somatic cell reprogramming. A yield 100-fold higher than the four-factor method alone can be attained through application of the DNA methyltransferase inhibitor, 5-azacytidine. A small molecule,70 BIX 01294, has a similar mechanism in that it enhances iPS cell conversion through inhibiting G9a histone methyltransferase, particularly when used in combination with Bay K 8644.71
To reduce the number of transcription factors used in reprogramming to just two (Oct4 and Sox2), this combination of small molecules can be used. Furthermore, combinations of small molecules have also been proven to induce iPS cell reprogramming in the absence of all of the transcription factors from the four-factor-method.72 For small molecules, another target in the reprogramming of somatic cells is the TGF-β pathway, as in the generation of both mouse and human iPS cells, inhibitors of the pathway have been used. While RepSox is a selective inhibitor of TGF-βR1, a 83-01 is an inhibitor of TGF-βR1, ALK4 and ALK7 activation. roles. In the generation of iPS cells, both compounds have roles.73
Conclusion
To conclude, within stem cell research, small molecules play important roles. This is because their application can regulate stem cell proliferation, or the small molecules can be used to move stem cell fate down specific differentiation lineages. In stem cell biology, their role is determined by their action on specific key signaling pathways. Thus, insight can be provided into the mechanisms of key signaling pathways by high-throughput screening and studying the mechanism of such molecules.
As we move towards a future wherein stem cell research is adequately understood and increasingly popular, the demand for small molecules that can modulate cell fate grows, in part because investigators need reliable molecular tools to continue to control stem cell fate and proliferation.
For stem cell therapy to become widely available, large populations of pluripotent stem cells must be maintained in culture, and the efficiency and reproducibility of guided cell differentiation must be maintained.
In the in vitro culture of such cells, small molecules play a significant role in their potential to enable guided differentiation for cell replacement therapies. Furthermore, small molecules modulating integral stem cell pathways may have a large role to play in the modulation of endogenous stem cell and progenitor cell populations. These small molecules may additionally encourage self-repair, through their promotion of the proliferation and differentiation of host cells, using pharmacological cues. Thus, this work done to develop and characterize small molecules which can potentially control self-renewal and differentiation is vital in advance of stem cell research: it seems likely that the research holds clues to its applications in both drug discovery and regenerative medicine.
References
- Ying et al. (2008) Nature 453 519
- Young (2011) Cell 144 940
- Bilic and Izpisua (2012) Belmonte Stem Cells 30 33
- Morrison and Scadden (2014) Nature 505 327
- Templeton et al. (2014) Cancer Stem Cells 1 1
- Lyssiotis et al. (2011) Angew.Chem.Int.Ed. 50 200
- Kumagai et al. (2013) Biochem.Biophys.Res.Comm. 434 710
- Maltman et al. (2009) Mol.Biosyst. 5 458
- Maden (2007) Nat.Rev.Neurosci. 8 755
- Rhinn and Dollé (2012) Development 139 843
- Tanoury et al. (2014) J.Cell Sci. 127 2095
- Han et al. (2003) Bioorg.Med.Chem. 11 3839
- Christie et al. (2008) Org.Biomol.Chem. 6 3497
- Christie et al. (2010) J.Neurosci.Methods 193 239
- Ingham and McMahon (2001) Genes Dev. 15 3059
- Taipale et al. (2002) Nature 418 829
- Kogerman et al. (1999) Nat.Cell.Biol. 1 312
- Binns et al. (1959) J.Am.Vet.Med.Assoc. 134 180
- Chen et al. (2002) Proc.Natl.Acad.Sci.U.S.A. 99 14071
- Epstein (2008) Nat.Rev.Cancer 8 743
- Lauth et al. (2007) Proc.Natl.Acad.Sci.U.S.A. 104 8455
- Chen et al. (2002) Proc.Natl.Acad.Sci.U.S.A. 99 14071
- Sinha and Chen (2006) Nat.Chem.Biol. 2 29
- Wu et al. (2002) J.Am.Chem.Soc. 124 14520
- Stanton et al. (2009) Nat.Chem.Biol. 5 154
- Trinh et al. (2014) Med.Chem.Commun. 5 117
- Lee et al. (2007) Chem.Biochem. 8 1916
- Derynck and Zhang (2003) Nature 425 577
- Wu and Hill (2009) Dev.Cell 16 329
- Hong and Yu (2009) Cytokine Growth Factor Rev. 20 409
- Kaminska et al. (2005) Acta.Biochim.Pol. 25 329
- Watabe et al. (2003) J.Cell.Biol. 163 1303
- Golestaneh and Mishra (2005) Oncogene 24 5722
- Ichida et al. (2009) Cell Stem Cell 5 491
- Willems et al. (2012) Cell Stem Cell 11 242
- Hao et al. (2008) PLoS One 3 e2904
- Borowiak et al. (2009) Cell Stem Cell 4 348
- Reya and Clevers (2005) Nature 434 843
- Katoh and Katoh (2007) Clin.Cancer Res. 13 4042
- Chen et al. (2009) Nat.Chem.Biol. 5 100
- Huang et al. (2009) Nature 461 614
- Lepourcelet et al. (2004) Cancer Cell 5 91
- Doble and Woodgett (2003) J.Cell Sci. 116 1175
- Zhong et al. (2009) Mol.Biosyst. 5 1356
- Shao et al. (2005) J.Biol.Chem. 280 26565
- Gotoh (2009) Curr.Stem Cell Res.Ther. 4 9
- Thisse and Thisse (2005) Dev.Biol. 287 390
- Xu et al. (2005) Stem Cells 23 315
- Dailey et al. (2005) Cytokine Growth Factor Rev. 16 233
- Bansal et al. (2003) J.Neurosci.Res. 74 486
- Zhou et al. (2010) Chem.Biol. 17 285
- Batley et al. (1998) Life Sci. 62 143
- Rinehart et al. (2004) J.Clin.Oncol. 22 4456
- Silva et al. (2008) PLoS Biol. 6 e253
- Weinmaster and Kopan (2006) Development 133 3277
- Kanungo et al. (2008) J.Neurochem. 106 2236
- Cullion et al. (2009) Blood 113 6172
- Mallon et al. (2006) Int.J.Biochem.Cell Biol. 38 1063
- Xu et al. (2008) Nature 453 338
- Chen et al. (2006) Proc.Natl.Acad.Sci.U.S.A. 103 17266
- Miyabayashi et al. (2008) Biosci.Biotechnol.Biochem. 72 1242
- Sato et al. (2004) Nat.Med. 10 55
- Ding et al. (2003) Proc.Natl.Acad.Sci.U.S.A. 100 7632
- Miyabayashi et al. (2007) PNAS 104 5668
- Takahashi and Yamanaka (2006) Cell 126 663
- Chen et al. (2004) J.Am.Chem.Soc. 126 410
- Anastasia et al. (2006) Cell Death Differ. 13 2042
- Huangfu et al. (2008) Nat.Biotechnol. 26 795
- Huangfu et al. (2008) Nat.Biotechnol. 26 1269
- Tsuji-Takayama et al. (2004) Biochem.Biophys.Res.Commun. 323 86
- Shi et al. (2008) Cell Stem Cell 3 568
- Hou et al. (2013) Science 341 651
- Ichida et al. (2009) Cell Stem Cell 5 49
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.