The three human RAS genes (NRAS, KRAS, and HRAS) encode four highly related RAS small GTPases (NRAS, KRAS4A, KRAS4B, and HRAS).1,2 RAS proteins operate as GDP-GTP regulated binary on-off switches which modulate a heterogeneous network of cytoplasmic signaling networks.3 Among cancer and developmental disorders (RASopathies), mutationally activated RAS proteins are the driving force behind abnormal signal transduction.4 RAS proteins are also the founding members of a substantial superfamily of small GTPases, comprising over 150 members.5
RAS genes constitute the most regularly mutated oncogene family in cancer. Indeed, RAS genes constituted the most regularly mutated oncogenes in the top three causes of cancer deaths in the US in 2016 (colorectal, lung, and pancreatic cancers).6 As a result, there has been an intensity of effort and interest focused on targeting RAS for cancer treatment.7
This review analyzes the weaknesses of RAS which have undergone exploitation for the development of pharmacologic inhibitors of RAS function, specifically focusing on RAS-dependent metabolic processes. These inhibitors also represent useful research reagents with which to examine RAS function. Since mutant RAS proteins embody cell context divergences in function, this analysis will focus on the example of pancreatic ductal adenocarcinoma (PDAC), arguably cancer most addicted to mutant RAS.8
RAS Mutations in Cancer
Altogether, the three RAS genes undergo mutational activation in ~25% of all cancers (COSMIC v80). Nonetheless, the frequency of RAS missense mutations is not homogeneous among cancer types. For instance, RAS genes are rarely mutated in breast cancers (<2%). In contrast, PDAC, the chief form of pancreatic cancer, displays the highest frequency (95%) of RAS mutations. Furthermore, the frequency of missense mutations located in each RAS gene is not homogeneous, with KRAS constituting 85% of all RAS mutations in cancer, followed by NRAS (12%), and, irregularly, HRAS (3%) (Figure 1A).1
While NRAS is the principal isoform mutated in melanoma and acute myelogenous leukemia (AML), KRAS is the principal RAS isoform mutated in PDAC, colorectal (CRC) and lung adenocarcinoma (LAC). While generally rare, HRAS mutations are principal in bladder and head and neck squamous cell carcinomas (HNSCC). RAS mutations play well-defined parts in the development of divergent cancers. In CRC, mutational loss of the APC tumor suppressor gene represents the commencing stage, with ensuing KRAS mutation promoting tumor progression.9 Contrastingly, in PDAC, KRAS mutations are the commencing genetic modification, with just under 95% of the earliest precancerous lesions, pancreatic intraepithelial neoplasias (PanINs), embodying KRAS mutations (Figure 1B).10
Further genetic modifications are needed for progression to the fully invasive and metastatic disease. Among genetically engineered mouse models of cancer, Kras mutation is the singular driving force behind the development of PanIN lesions, with fully malignant cancers only arising rarely and after substantial time.11 Nonetheless, synchronous loss of one of the three tumor suppressor gene mutations commonly lost in human PDAC (Cdkn2a, Smad4, and Tp53) significantly increases and accelerates the high occurrence of metastatic disease.12–14
In spite of the initial onset of KRAS mutations in PDAC development, as well as the subsequent accumulation of over 50 additional gene mutations in advanced cancers,15–20 there is extensive evidence from cell culture and mouse models indicating how persistent mutant KRAS function is crucial for the maintenance of malignant disease.21–25 Genetic ablation of mutant KRAS engenders fast tumor regression, demonstrating the possible effects of effective KRAS treatment. Offsetting the clear therapeutic advantage in targeting KRAS is proof that cancers possess the capacity to adapt and acquire mechanisms (such as YAP1 activation) that overcome their addiction to mutant KRAS.26,27
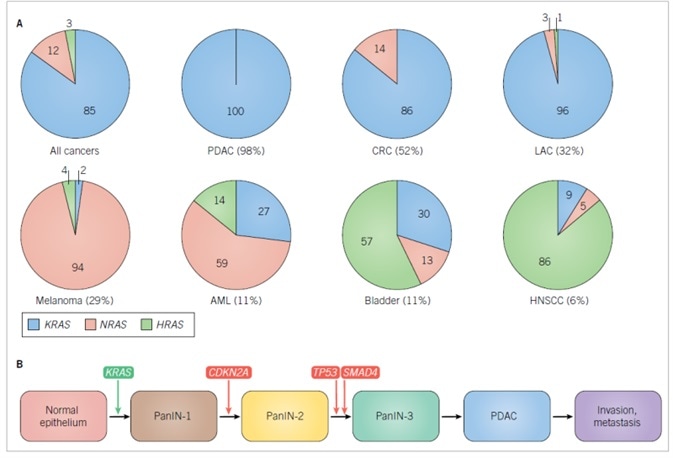
Figure 1. RAS isoform mutations in cancer.
A. Frequencies of RAS missense mutations in different cancer types. The numbers in parentheses are the percentage of all cancers harboring RAS mutations. The numbers in the pie charts indicate the percentage of mutations in each RAS isoform. Data were compiled from Supplementary Table 2 found in Reference 1. Abbreviations used are: AML, acute myelogenous leukemia; CRC, colorectal cancer; HNSCC, head, and neck squamous cell carcinoma; LAC, lung adenocarcinoma; PDAC, pancreatic ductal adenocarcinoma. B. Genes involved in the initiation and progress of PDAC. CDKN2A encodes two distinct tumor suppressor proteins, p16INK4A and p19ARF, that act as positive regulators of the RB and p53 tumor suppressors, respectively. SMAD4 encodes the SMAD4 transcription factor that is activated by TGFβ signaling.
RAS Protein Function
The three RAS genes encode particularly identical (82–90%) 188–189 amino acid proteins (Figure 2). KRAS encodes two splice variants due to usage of alternative exon 4 sequences, classified KRAS4A and 4B, with 4A more tissue restricted and 4B more broadly expressed. The N-terminal 164 residues of RAS proteins constitute the G domain (92–98% sequence identity) implicated in GTP binding and hydrolysis. The C-terminal 24/25 residues display the greatest difference between the divergent isoforms and comprise sequences that enable membrane interactions.
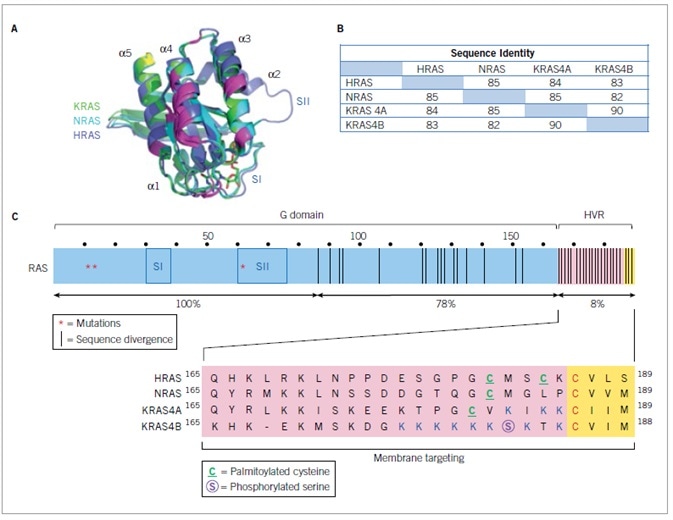
Figure 2. Human RAS proteins.
A. RAS protein structures. Overlay of the structures of the three RAS proteins. B. RAS isoform sequence identity. Percent sequence identity was determined by CLUSTALW multiple sequence alignment analysis. C. The three RAS genes encode four RAS protein isoforms that are highly identical in primary sequence (82–90% amino acid (aa) sequence identity), structure, and biochemical properties. The N-terminal 164 residues comprise the G domain, which is involved in GTP binding and hydrolysis and effector interaction. Within the G domain is the switch I (SI; residues 30-38) and II (SII; residues 60–76) regions, that change in conformation during GDP-GTP cycling. The C-terminal membrane targeting sequences are comprised of the hypervariable regions (HVR; residues 165–184/5), that share little sequence identity (8%), followed by the CAAX tetrapeptide sequences that signal for post-translational modifications required to promote RAS membrane association.
RAS GDP-GTP Regulation
Wild-type (WT) RAS proteins display low picomolar affinity for GDP and GTP and behave as GDP-GTP modulated binary on-off switches (Figure 3A).3 Among quiescent cells, WT RAS proteins exist in their inactive GDP-bound state. Following stimulation of cell surface receptors (such as EGFR) by a broad range of extracellular stimuli, activation of cytoplasmic signaling components subsequently activates RAS-selective guanine nucleotide exchange factors (RASGEFs; e.g., SOS1). This, in turn, stimulates transient and rapid GDP-GTP exchange and formation of the active GTP-bound protein. The active state is quickly ended by GTPase activating proteins (RASGAPs; e.g., neurofibromin), stimulating the weak intrinsic GTPase activity of RAS, which hydrolyzes the bound GTP to GDP, and returns RAS to the inactive state.
Throughout GDP-GTP cycling, RAS protein conformation is modified at the switch I (SI) and II (SII) regions (Figures 2A), with the conformation in the GTP-bound state exhibiting augmented affinity for the RAS-binding (RBD) and RAS-association domains located on RAS effectors (Figure 3).28 Mutated RAS genes located in cancer embody singular missense mutations which are found principally (98–99%) at one of three mutational hotspots, glutamine-61 (Q61), glycine-12 (G12) and glycine-13 (G13) (Figure 2C).29,30
The consequent singular amino acid substitutions impede intrinsic and GAP-stimulated GTP hydrolysis activity, engendering mutant proteins that are continually GTP-bound and active without external stimuli (Figure 3B). WT RAS proteins might also be activated persistently in cancer via indirect mechanisms. Most notably, loss of function of the RASGAP, neurofibromin (encoded by the NF1 tumor suppressor gene), is triggered by cancer-associated mutations (Figure 3B).31,32 Mutational activation or overexpression of cell surface receptor tyrosine kinases (RTKs; e.g., EGFR) may also engender persistent RAS activation via activation of RASGEFs. Amplification of WT RAS genes is also observed in cancer (Figure 3D).
RAS Effectors
The RBD/RA domains are connected to an array of catalytically diverse effectors (Figure 4), with RAF serine/threonine kinases (ARAF, BRAF, and CRAF/RAF1, ) the best-validated effectors significant for driving the oncogenic function of mutant RAS proteins.33 RAS-GTP binds and promotes RAF trafficking to the inner face of the plasma membrane, where additional events (such as protein kinase-mediated phosphorylation) promote activation of RAF catalytic activity. This leads to phosphorylation of the MEK1 and MEK2 dual-specificity protein kinases.
Activation of MEK1/2 subsequently phosphorylates and activates the ERK1 and ERK2 mitogen-activated protein kinases (MAPKs). Activated ERK1/2 is able to phosphorylate up to 200 cytoplasmic and nuclear substrates. The MYC transcription factor is one of the key ERK substrates important in RAS-driven cancer growth. The significance of the RAF effector pathway in RAS oncogenic function is reinforced by the regular mutational activation of BRAF (19%; COSMIC v79) in cancer, with generally mutually exclusive incidence among cancers embodying RAS mutations.
The second best-validated RAS effector family comprises (p110a/g/d) catalytic subunits of the heterodimeric class I phosphoinositide-3-kinases (PI 3-K).34 These lipid kinases phosphorylate phosphoinositide (4,5) bisphosphate (PIP2) to produce the membrane-associated lipid second messenger phosphoinositide (3,4,5) trisphosphate (PIP3). PIP3 is bound by pleckstrin homology (PH) domain-containing proteins, particularly the AKT serine/threonine kinases, thus promoting AKT activation and engendering activation of the mTORC1 complex.
The significance of the PI 3-K effector pathway in oncogenesis is underpinned by the regular mutational activation of PIK3CA (10%; COSMIC v79) in cancer, even though the coincidence of these mutations with RAS mutations suggests that mutated RAS alone might not always cause potent activation of PI 3-K signaling.
The RALGEF-RAL pathway represents the third best-validated RAS effector.35,36 RALGEFs are Ras-association (RA) domain-containing proteins acting as GEFs and activators of the RALA and RALB RAS-like small GTPases. While elements of this pathway do not undergo mutation in cancer, extensive evidence from cell culture and mouse models reveals the necessity of both RALB and RALA in RAS-dependent cancer growth. Notably, in spite of their robust sequence and biochemical identity, RALB and RALA display well-defined functions in this process. For instance, while RALA was needed for PDAC tumorigenic growth, whereas RALB was not, RALB was essential for PDAC invasion and metastasis.37
The fourth best-validated RAS effector pathway comprises RACGEF activation of the RAC small GTPase.1 The contemporary discovery of mutationally activated RAC1 mutants in melanoma reinforces the significant part played by this effector pathway in cancer growth.38 Mouse model examinations revealed that genetic ablation of RAC1 impaired mutant RAS-driven cancer growth.39,40
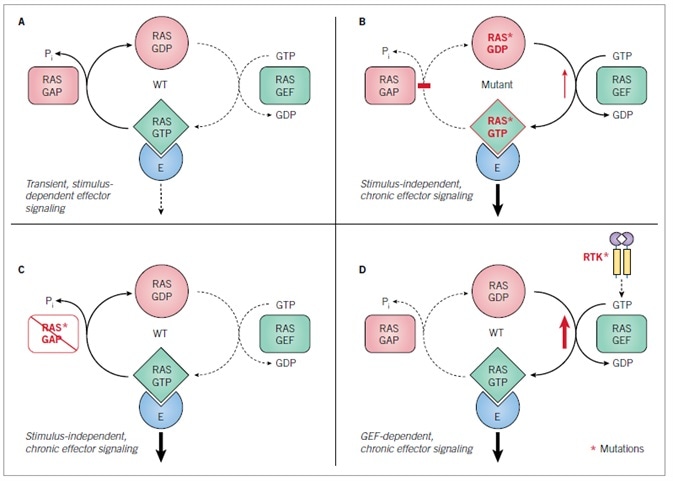
Figure 3. RAS GDP-GTP cycle.
A. Wild type RAS protein GDP-GTP regulation. B. Mutant RAS proteins with single amino acid substitutions at residues G12, G13 or Q61 have impaired GTPase activity and/or enhanced nucleotide exchange. C. The loss of RASGAP function causes constitutive activation of wild type RAS. D. Aberrant receptor tyrosine kinase signaling causes RASGEF-dependent activation of wild type RAS.
RAS Membrane Association
RAS protein function critically depends on association with the inner face of the plasma membrane.29,41 RAS proteins undergo initial synthesis as cytosolic, inactive proteins (Figure 5). They are subsequently subject to rapid post-translational alterations that promote RAS association with the plasma membrane.
The carboxyl-terminal CAAX tetrapeptide motif (C = cysteine, A = aliphatic amino acid, X = terminal amino acid) indicates the first alteration, the covalent addition of a C15 farnesyl isoprenoid lipid, catalyzed by the cytosolic enzyme farnesyltransferase (FTase). This alteration is attended by subsequent endoplasmic reticulum (ER) associated RASconverting enzyme 1 (RCE1) catalyzed proteolytic removal of the AAX residues, which is, in turn, followed carboxylmethylation of the now terminal farnesylated cysteine by isoprenylcysteine methyltransferase (ICMT). The essential role of these alterations for RAS function is shown by mutation of the Cys residue to Ser (CAAX > SAAX), which prevents all three alterations: SAAX mutants of RAS proteins embodying oncogenic mutations are cytosolic and totally inhibited in oncogenic function.
While the CAAX-signaled alterations are critical for RAS operation, further carboxyl-terminal sequence elements are required for adequate RAS subcellular localization and membrane association.29,41 NRAS, KRAS4A and HRAS comprise cysteine residues immediately upstream of the CAAX motif which is sites of post-translational alteration by C16 fatty acid palmitate moieties. The palmitoyl acyltransferases (PATs) implicated in catalyzing palmitate covalent attachment are complex and still not entirely established for RAS.
Moreover, in contrast to the irreversible and universal farnesyl alteration, palmitate alteration is connected to only a small proportion of RAS, which may undergo reversible deacylation by an acyl protein thioesterase (APT1/2). Therapy with the reasonably non-specific PAT inhibitor 2-bromopalmitate disturbs the plasma membrane association of palmitoylated RAS isoforms.
The acylation/deacylation cycle of RAS proteins is importantly implicated in RAS cycling between the plasma membrane, cytoplasm, and Golgi membranes. Differential RAS signaling at distinctive subcellular compartments is also characterized by complexity, and its delineation is ongoing. KRAS4B does not embody any palmitate-modified cysteine residues (Figure 2C).29,41 Rather, a lysine-rich polybasic domain alongside the CAAX motif includes a second membrane-targeting sequence element which, in conjunction with the CAAX alterations, promotes KRAS membrane association and oncogenic activity (Figure 5).
The positive charge supplied by this polybasic sequence enables plasma membrane association, partially by association with negatively charged phospholipids. The KRAS4A splice variant, which embodies a more restricted tissue expression profile, also comprises carboxyl-terminal lysine-rich sequences which, in conjunction with a palmitoylated cysteine, are necessitated for complete KRAS4A membrane association and function.42 The KRAS4B polybasic domain comprises a serine residue which can undergo phosphorylation by protein kinase C (Figure 2C).43 This reversible phosphorylation event modifies KRAS4B subcellular localization, promoting association with endomembranes, and engendering a loss of KRAS4B oncogenic function.44 Therapy with bryostatin, an activator of protein kinase C, promotes KRAS4B phosphorylation. The phosphorylation status of this serine residue also governs KRAS4B association with calmodulin.
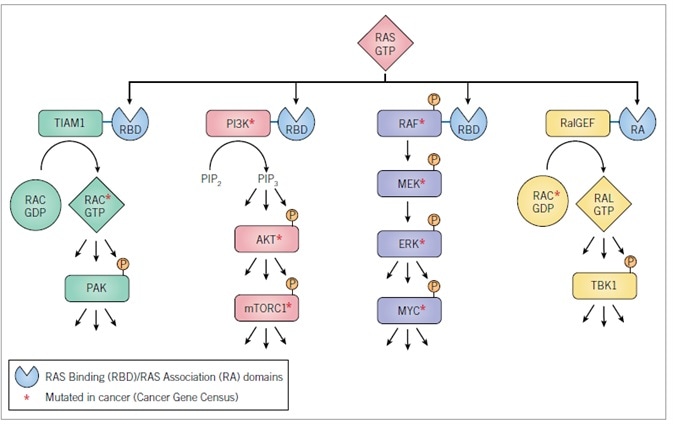
Figure 4. RAS effectors
Effectors validated to drive mutant RAS-dependent cancer growth. Cell culture and mouse model analyses have validated the requirement for specific components of these effector pathways in the maintenance and/or initiation and progression of tumor growth. Activated RASGTP recognizes RBD or RA domain-containing proteins.
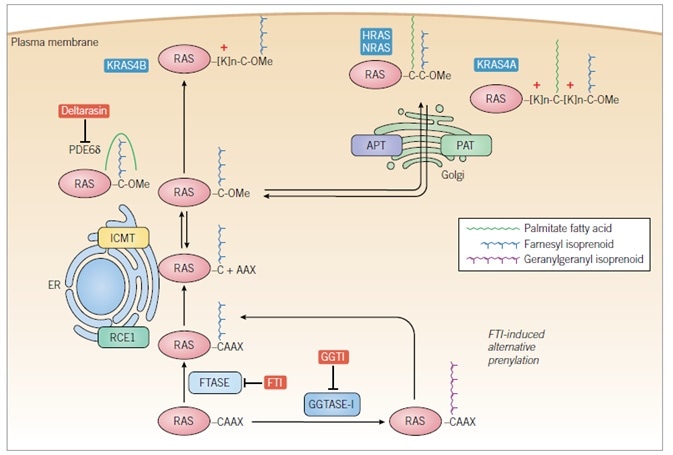
Figure 5. Regulation of RAS subcellular localization and membrane association
RAS proteins are synthesized initially as cytosolic proteins. Within minutes, the CAAX motif signals for cytosolic FTase-catalyzed covalent and irreversible addition of a C15 isoprenoid to the cysteine residue. The endoplasmic reticulum (ER)-associated RAS-converting enzyme 1 (RCE1) then catalyzes endoproteolytic removal of the AAX residues, followed by ER-associated isoprenylcysteine carboxylmethyltransferase (ICMT) catalysis of reversible methyl esterification of the now terminal farnesylated cysteine residue using S-adenosylmethionine as the methyl donor. The CAAX-signaled modifications comprise the first signal that is necessary but not sufficient to promote plasma membrane association and subcellular localization essential for RAS protein function. The second signal is contributed either by polybasic sequences (KRAS4A, KRAS4B) and/or by cysteine residues (HRAS, NRAS, and KRAS4A) that are reversibly covalently modified by Golgi-associated palmitoyl acyltransferase (PAT)-catalyzed addition of a C16 palmitoyl fatty acid. While one PAT (DHHC9-GPC16) has been identified for HRAS, whether other PATs for RAS proteins exist, and the acyl-protein thioesterases (APT) that depalmitoylate RAS proteins remain poorly understood. PDE6d recognizes the farnesyl group of KRAS4B and solubilizes the protein from the ER, promoting availability for the plasma membrane; this activity can be blocked by deltarasin and other small molecules. FTIs block the first CAAX-signaled modification, preventing all subsequent modifications, rendering RAS cytosolic and inactive. In the absence of FTase activity, KRAS and NRAS are substrates for GGTase-I and alternative prenylation by covalent addition of a C20 geranylgeranyl isoprenoid, allowing RCE1 and ICMT modification and plasma membrane association. Concurrent treatment with FTI and GGTI can block KRAS and NRAS membrane association.
Produced from materials originally authored by Kirsten L. Bryant, Adrienne D. Cox and Channing J. Der from University of North Carolina at Chapel Hill, Lineberger Comprehensive Cancer Center, Department of Pharmacology Department of Radiation Oncology, Chapel Hill, NC 27599, USA.
References and Further Reading
- Cox et al. (2014) Nat Rev Drug Discov 13 828.
- Stephen et al. (2014) Cancer Cell 25 272.
- Cox et al. (2010) Small GTPases 1 2.
- Rauen. (2013) Annu Rev Genomics Hum Genet 14 355.
- Wennerberg et al. (2005) J Cell Sci 118 843.
- Siegel et al. (2016) CA Cancer J Clin 66 7.
- Ledford. (2015) Nature 520 278.
- Bryant et al. (2014) Trends Biochem Sci 39 91.
- Fearon. (2011) Annu Rev Pathol 6 479.
- Ryan et al. (2014) N Engl J Med 371 2140.
- Hingorani et al. (2003) Cancer Cell 4 437.
- Aguirre et al. (2003) Genes Dev 17 3112.
- Hingorani et al. (2005) Cancer Cell 7 469.
- Kojima et al. (2007) Cancer Res 67 8121.
- Jones et al. (2008) Science 321 1801.
- Biankin et al. (2012) Nature 491 399.
- Sausen et al. (2015) Nat Commun 6 7686.
- Waddell et al. (2015) Nature 518 495.
- Witkiewicz et al. (2015) Nat Commun 6 6744.
- Bailey et al. (2016) Nature 531 47.
- Brummelkamp et al. (2002) Cancer Cell 2 243.
- Lim et al. (2008) Nature 452 646.
- Collins et al. (2012) J Clin Invest 122 639.
- Collins et al. (2012) PLoS One 7 e49707.
- Ying et al. (2012) Cell 149 656.
- Kapoor et al. (2014) Cell 158 185.
- Shao et al. (2014) Cell 158 171.
- Vetter. in Ras superfamily small G proteins: biology and Mechanisms 1 Vol. 1 (ed A. Wittinghofer) Ch. 2, 25 (Spinger-Verlag Wien, 2014).
- Cox et al. (2015) Clin Cancer Res 21 1819.
- Prior et al. (2012) Cancer Res 72 2457.
- Bos et al. (2007) Cell 129 865.
- Vigil et al. (2010) Nat Rev Cancer 10 842.
- Ryan et al. (2015) Trends Cancer 1 183.
- Fritsch et al. (2013) Cell 154 940.
- Bodemann et al. (2008) Nat Rev Cancer 8 133.
- Gentry et al. (2014) Biochim Biophys Acta 1843 2976.
- Lim et al. (2006) Curr Biol 16 2385.
- Halaban. (2015) Clin Ther 37 682.
- Heid et al. (2011) Gastroenterology 141 719.
- Kissil et al. (2007) Cancer Res 67 8089.
- Ahearn et al. (2012) Nat Rev Mol Cell Biol 13 39.
- Tsai et al. (2015) Proc Natl Acad Sci U S A 112 779.
- Bivona et al. (2006) Mol Cell 21 481.
- Sung et al. (2013) Proc Natl Acad Sci U S A 110 20593.
- Marcus et al. (2015) Clin Cancer Res 21 1810.
- Ostrem et al. (2016) Nat Rev Drug Discov 15 771.
- Welsch et al. (2017) Cell 168 878.
- Ostrem et al. (2013) Nature 503 548.
- Lim et al. (2014) Angew Chem Int Ed Engl 53 199.
- Lito et al. (2016) Science 351 604.
- Patricelli et al. (2016) Cancer Discov 6 316.
- Waldmann et al. (2004) Angew Chem Int Ed Engl 43 454.
- Muller et al. (2004) Angew Chem Int Ed Engl 43 450.
- Karaguni et al. (2002) Cancer Res 62 1718.
- Herrmann et al. (1998) Oncogene 17 1769.
- Pan et al. (2008) Cell Signal 20 1134.
- Berndt et al. (2011) Nat Rev Cancer 11 775.
- Marom et al. (1995) J Biol Chem 270 22263.
- Blum et al. (2008) Recent Pat Anticancer Drug Discov 3 31.
- Papke et al. (2016) Nat Commun 7 11360.
- Zimmermann et al. (2013) Nature 497 638.
- Martin-Gago et al. (2017) Angew Chem Int Ed Engl 56 2423.
- Cho et al. (2015) Mol Cell Biol 36 363.
- van der Hoeven et al. (2013) Mol Cell Biol 33 237.
- Foster et al. (2016) Cancer Cell 29 477.
- Chen et al. (2016) Cancer Discov 6 300.
- Collisson et al. (2012) Cancer Discov 2 685.
- Samatar et al. (2014) Nat Rev Drug Discov 13 928.
- . Peng et al. (2014) Cancer Res 74 Abstract nr DDT02.
- Zhang et al. (2015) Nature 526 583.
- Morris et al. (2013) Cancer Discov 3 742.
- Fruman et al. (2014) Nat Rev Drug Discov 13 140.
- Baker et al. (2014) Clin Cancer Res 20 4740.
- Athuluri-Divakar et al. (2016) Cell 165 643.
- Ritt et al. (2016) Mol Cell 64 875.
- Downward. (2015) Clin Cancer Res 21 1802.
- Barbie et al. (2009) Nature 462 108.
- Luo et al. (2009) Cell 137 835.
- Scholl et al. (2009) Cell 137 821.
- Yu et al. (2013) Enzymes 34 Pt. B 201.
- Wang et al. (2017) Cell.
- White. (2013) Genes Dev 27 2065.
- Kimmelman. (2015) Clin Cancer Res 21 1828.
- Warburg. (1956) Science 123 309.
- Liberti et al. (2016) Trends Biochem Sci 41 211.
- Hanahan et al. (2011) Cell 144 646.
- Racker et al. (1985) Proc Natl Acad Sci U S A 82 3535.
- Yun et al. (2009) Science 325 1555.
- Le et al. (2010) Proc Natl Acad Sci U S A 107 2037.
- Rajeshkumar et al. (2015) Cancer Res 75 3355.
- Boudreau et al. (2016) Nat Chem Biol 12 779.
- Amin et al. (2016) Gut Liver 10 665.
- Reni et al. (2016) Clin Cancer Res 22 1076.
- Kordes et al. (2015) Lancet Oncol 16 839.
- Cameron et al. (1978) Proc Natl Acad Sci U S A 75 4538.
- Moertel et al. (1985) N Engl J Med 312 137.
- Creagan et al. (1979) N Engl J Med 301 687.
- Yun et al. (2015) Science 350 1391.
- Wise et al. (2010) Trends Biochem Sci 35 427.
- Daye et al. (2012) Semin Cell Dev Biol 23 362.
- Hosios et al. (2016) Dev Cell 36 540.
- Son et al. (2013) Nature 496 101.
- Wang et al. (2010) Cancer Cell 18 207.
- Robinson et al. (2007) Biochem J 406 407.
- Chakrabarti et al. (2015) Cancer Metab 3 12.
- Ha et al. (2016) Front Physiol 7 381.
- Bar-Sagi et al. (1986) Science 233 1061.
- Commisso et al. (2013) Nature 497 633.
- Davidson et al. (2017) Nat Med 23 235.
- Palm et al. (2015) Cell 162 259.
- Von Hoff et al. (2013) N Engl J Med 369 1691.
- Von Hoff et al. (2011) J Clin Oncol 29 4548.
- Yang et al. (2011) Genes Dev 25 717.
- Guo et al. (2011) Genes Dev 25 460.
- Elgendy et al. (2011) Mol Cell 42 23.
- Young et al. (2009) Genes Dev 23 798.
- Rosenfeldt et al. (2013) Nature 504 296.
- Yang et al. (2014) Cancer Discov 4 905.
- Sousa et al. (2016) Nature 536 479.
- Wolpin et al. (2014) Oncologist 19 637.
- Boone et al. (2015) Ann Surg Oncol 22 4402.
- Liu et al. (2011) Cell 147 223.
- Egan et al. (2015) Mol Cell 59 285.
- Petherick et al. (2015) J Biol Chem 290 11376.
About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.