It is well known that through the interplay of hormonal and neural mechanisms, the expression of appetite is chemically coded in the hypothalamus.1 In brief, it is proposed that the hypothalamus houses opposing sets of neuronal circuitry: an appetite-inhibitory circuit and an appetite-stimulatory circuit.2
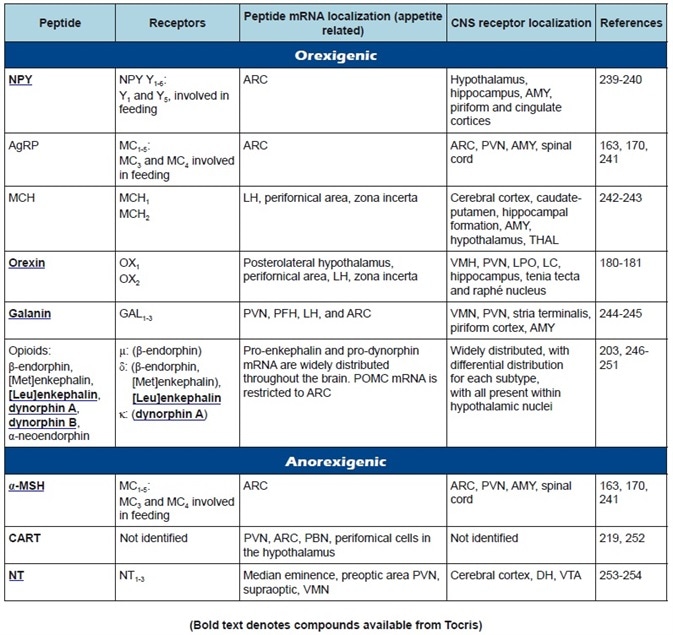
Table 1a. Central appetite regulatory peptides: receptor classification, peptide, and receptor localization.
These circuits are affected by afferent neural and peripheral hormonal signals which give feedback and integrative processing of energy intake, nutritional status, and expenditure. The anorexigenic neurotransmitters released by the inhibitory circuit decrease appetite, while the appetite-stimulatory circuit expresses orexigenic neurotransmitters which promote appetite.
Further to modulation by signals originating in the periphery, these integrative functions are influenced by a large scope of neural influences within the brain, cognitive, reflecting sensory, memory, and affective processes.
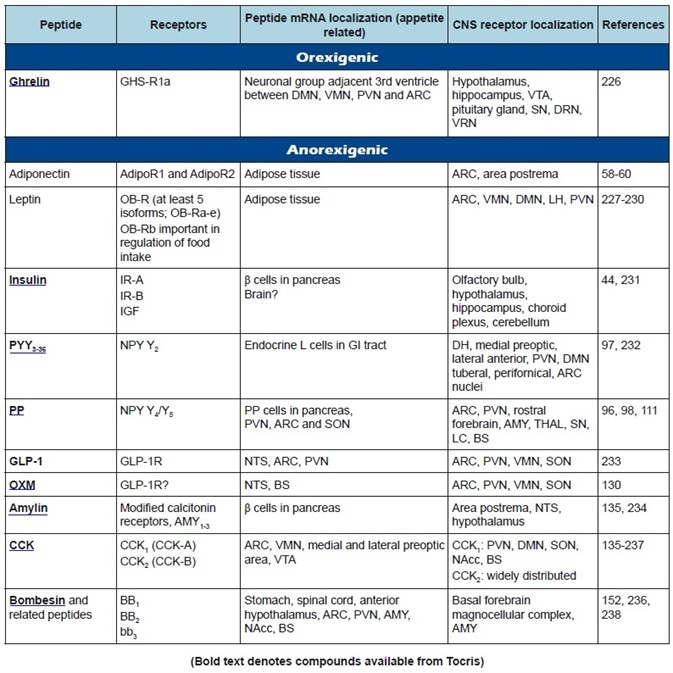
Table 1b. Peripheral appetite regulatory peptides: receptor classification, peptide, and receptor localization.
This article gives an outline of the increasing amount of peptides that have been implicated in energy homeostasis and appetite regulation and describes the putative roles of most of the currently known participants. The extensive literature on the physiological control of food intake, body weight, and metabolism regulation is talked about in more depth in some recent publications.3-11
It should be known that for this article, appetite-modulating peptides are considered in terms of their central or peripheral origins and actions; yet, most peptides that were originally considered to be exclusively synthesized in the periphery are now also known to be generated in the central nervous system (CNS).
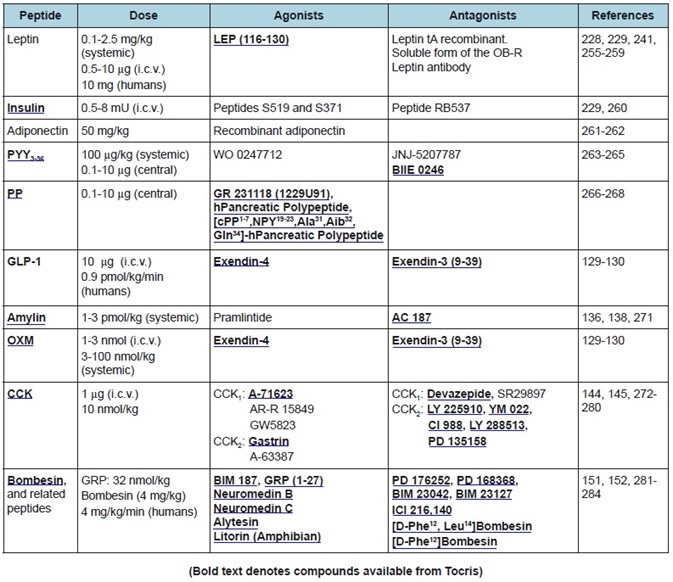
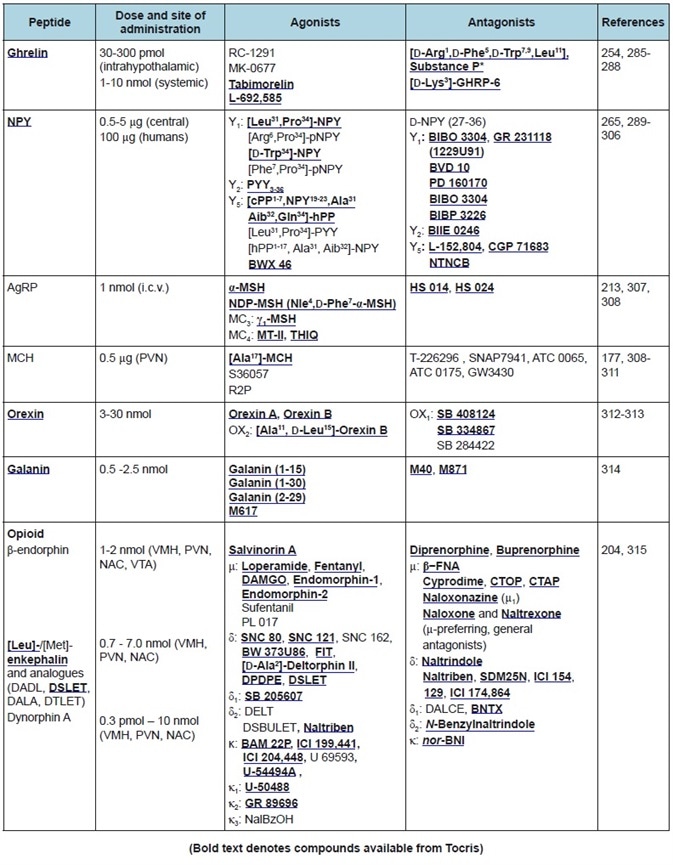
A summary of the featured peptides, plus their receptors and anatomical distributions, can be found in Table 1. Tables 2-4 show the commonly employed doses, antagonists, and agonists of the peptides discussed.
Peripheral Peptides Regulating Appetite
Leptin
Leptin is a 146 amino acid, glycosylated protein. This adipokine is predominantly produced by white adipose tissue, but low levels of expression are also detected in the hypothalamus.12,13 Since it was identified as the product of the ob gene, it has been central to the investigation of body weight regulation and appetite. Genetic mutation of this gene can be seen in phenotypically hyperphagic, leptin-deficient, and obese ob/ob mice.
Additionally, mutations in the leptin receptor gene are associated with obesity in db/db mice and fa/fa rats. In animals and humans, circulating leptin levels are directly related to the amount and size of adipocytes, and so better correspond with total fat mass than with bodyweight.14
It has been proposed that leptin conveys information to the hypothalamus concerning the amount of energy stored in adipose tissue. Heightened levels are suggested to suppress appetite and influence energy expenditure to regulate body weight. Administration of leptin has been found to decrease food intake in all species studied to date,15 including rodents,18,19 humans,16 non-human primates,17 and sheep.20
Table 2. Peripheral anorexigenic peptides. Commonly used doses, agonists, and antagonists.

Furthermore, microinjections of leptin in rodents, into the ventromedial hypothalamus (VMH)21 and the arcuate nucleus (ARC)22 can decrease food intake massively, suggesting that leptin’s actions are mediated mainly by the hypothalamus. Activation of these brain regions by leptin is partly attributable to its actions on ARC neurons which lie outside of the blood-brain barrier.23 Yet, active transport of leptin across the blood-brain barrier has been seen.24,25
Leptin responsive neurons in the ARC include those containing the agouti-related peptide (AgRP) and orexigenic peptides neuropeptide Y (NPY), and those containing the anorexigenic peptides α-melanocyte-stimulating hormone (α-MSH) and cocaine and amphetamine-regulated transcript (CART).
While α- MSH/CART neurons are activated, the NPY/AgRP neurons are blocked by leptin.26 There are also potentially synergistic interactions between the short-term satiety signal cholecystokinin (CCK) and leptin, which may involve integration at the level of primary sensory afferents.27
Circulating leptin levels also differ in an adiposity-independent manner; becoming lower during fasting and heightening with re-feeding. These alterations have been linked to glucose and insulin regulation. For example, insulin enhances leptin production and plasma levels of leptin are correlated with plasma glucose levels.28-30
As its effects on food intake are transient, it has been said that the effect of leptin on energy expenditure could be the most prominent in terms of body weight regulation.31 One way that leptin enhances energy expenditure is via sympathetic activation of brown adipocytes, resulting in thermogenesis in brown adipose tissue (BAT).32 The effects of leptin on thermogenesis are also displayed in non-rodent species with comparatively low levels of BAT.
In sheep, the central administration of leptin markedly enhances postprandial thermogenesis in both muscle and diffuse adipose depots (retroperitoneal and gluteal fat).33 As previously discussed, genetic mutations leading to leptin receptor deficiencies or leptin insufficiency support the idea that this peptide plays a crucial role in long-term energy homeostasis.
Although several studies have stated that leptin can be an effective pharmaceutical tool for treating obesity in leptin-deficient states, the administration of exogenous leptin does not significantly decrease adiposity in the majority of instances of human obesity. Additionally, deficiencies in leptin receptor expression or leptin production have been associated with just a few instances of human obesity.34
Increased adipocyte leptin content and high circulating leptin levels are usually in the obese, which has led to the suggestion of leptin resistance. This hypothesis outlines the failure of an upregulated leptin signal to alter appetite and prevent weight gain. Leptin resistance looks to be due to a reduction in its transport across the blood-brain barrier, in addition to its lowered ability to initiate cellular activation within the brain.15
Leptin enters the brain via active transport, which involves a short form of the leptin receptor (ObRa) at the choroid plexus. Studies in rodents have shown that feeding animals a high-fat diet lower ObRa levels within the hypothalamus24,35 and, consistent with this, leptin transport is decreased in obese humans.36
Another cause could be a defect in leptin signaling which is related to the suppressor of cytokine signaling 3 (SOCS3) and insulin receptor substrate/phosphatidylinositol 3-kinase (IRS/PI 3-K) signaling pathways.37,38
Numerous studies have shown the significance of SOCS3 in establishing the degree of leptin sensitivity.39-41 For example, a particular increase in SOCS3 expression is observed in ARC neurons of mice with diet-induced obesity and this may be a key cause of leptin resistance.42
Insulin
Insulin is a 51 amino acid protein that is mainly produced by the pancreatic β cells in response to elevated blood glucose concentrations. There is also an indication of some neuronal synthesis, yet the majority of insulin in the brain is of peripheral origin.43,44 Circulating levels of insulin are proportional to adiposity, as with leptin.45 Insulin interacts with certain receptors in the hypothalamus46 and, along with leptin, regulates the synthesis and release of NPY.47
The expression of NPY in ARC neurons is lowered after central or systemic administration of leptin and insulin, but these NPY neurons are activated when the levels of these hormones decrease during undernutrition.48 Intrahypothalamic or intraventricular administration of insulin slows food intake and creates a sustained loss of body weight in both rodents49 and acts as a meal initiation signal. In primates, ghrelin levels fall.50
On the other hand, the injection of insulin antibodies into the hypothalamus of rats heightens food intake and leads to body weight gain.51 Also, mice with a genetic deletion of neuronal insulin receptors are obese and hyperphagic.52 Insulin secretion is acutely stimulated in response to meals. In a large majority of obese humans, obesity is associated with both hyperleptinemia and hyperinsulinemia, indicative of leptin, as well as insulin resistance.

Adiponectin
Adiponectin (also known as Acrp30 and apM1) is a 244 amino acid polypeptide which modulates a variety of metabolic processes, including fatty acid catabolism and glucose regulation. It is produced exclusively by mature adipocytes53 and levels are decreased in obesity, particularly visceral obesity.54
Via a peripheral mechanism, this is thought to contribute to lower insulin sensitivity and the development of insulin resistance.55 Although adiponectin does not seem to cross the blood-brain barrier,56,57 the ARC58 and the area postrema59 respond to adiponectin, suggesting that these cells could be involved in relaying the signal to other brain regions.
It is possible that the lower ghrelin levels in the obese may be a consequence of overconsumption, instead of the cause of the signal to other brain regions. In the hypothalamus, actions of adiponectin are mediated via two adiponectin receptors (AdipoR1 and AdipoR2), which have opposing effects.60
Deletion of the AdipoR1 gene leads to obesity caused by lower energy expenditure, whereas deletion of the AdipoR2 gene leads to higher energy expenditure and a lean phenotype.61
Central administration of adiponectin lowers body weight,62 mainly a result of a growth in energy expenditure. In wild-type and ob/ob mice, adiponectin heightens uncoupling protein 1 (UCP1, thermogenin) mRNA levels in BAT and encourages thermogenesis, without modifying food intake.62 To date, the effects of adiponectin on food intake have been inconclusive, with studies showing either a lack of effect,62 an increase,58 or a reduction.63
Adiponectin is a clearly important peripheral hormone for establishing levels of insulin sensitivity but more work is needed to resolve the actions of this hormone within the brain.
Ghrelin
Ghrelin is a 28 amino acid acylated peptide. It is endogenous for the growth hormone secretagogue ligand (GHS-R) and was the first circulating hormone shown to stimulate weight gain and eating. It is primarily secreted by specialized enterochromaffin cells located in the mucosa of the gastric fundus,64 although many studies have shown that it is also synthesized in the CNS, notably within hypothalamic regions.65,66
In lean humans, ghrelin levels are heightened during the time between meals (or when fasting) and peak before starting a meal, resulting in the idea that ghrelin could behave as a meal initiation signal. Ghrelin levels drop in the hour after a glucose load or a meal, with the extent of the postprandial suppression being proportional to caloric intake.
Notably, it has been reported that ghrelin infusion can increase food intake in healthy volunteers67 and in patients with anorexia due to cancer68 and chronic renal failure.69 Crucially, these effects happen at doses which are inside the normal physiological range for circulating endogenous ghrelin.
In humans, circulating ghrelin levels heightened in anorexia nervosa and during sustained fasting with weight loss70,71 and are lowered in acute states of positive energy balance and in obesity.
Individuals who are obese do not exhibit the postprandial decline in plasma concentrations observed in the lean, in addition to having lower ghrelin levels.72 It has been suggested that this lack of ghrelin suppression may lead to higher food consumption and add to the pathophysiology of obesity. Of course, ghrelin levels in obesity could already be lowered to a level where no further fall can be detected.

The metabolic effects of ghrelin are opposite to those of leptin and it has been suggested that these two peptides exert a counter-regulatory action on each other.73 Central and peripheral administration of ghrelin74,75 heightens carbohydrate metabolism and decreases energy expenditure and fat utilization in addition to possibly playing a part in the initiation of eating.74
By favoring an increase in glucose and triglyceride uptake, increasing lipogenesis, and stopping lipid oxidation in white adipocytes, central ghrelin seems to partition nutrients toward fat storage. This may also propose an alternative role for the pre-meal surge in ghrelin. Instead of being a signal of meal initiation, this increase can instigate processes in the CNS which prepare the body to receive and process incoming nutrients appropriately.
One mediator of the orexigenic effect of ghrelin is AMP-activated protein kinase (AMPK),76,77 a key enzyme regulator of energy homeostasis both peripherally and centrally.78,79 Furthermore, through stimulation of hypothalamic circuits, ghrelin looks to achieve its orexigenic action, in part by activating the arcuate NPY/AgRP pathways and opposing anorexigenic signals.80-82
Since feeding which is induced by intraparaventricular nuclear ghrelin is reversed by the CB1 receptor antagonist rimonabant, ghrelin-induced eating may also be mediated via the endogenous cannabinoid system.83
There are some inconsistencies in the data which indicate that caution should be shown, despite the huge interest in ghrelin and its putative role in stimulating eating. For example, it should be considered that, compared to other endogenous orexigens, ghrelin has only modest effects on food intake in animal models.
Also, ghrelin-deficient mice (ghrl–/–) display normal levels of hypothalamic orexigenic and anorexigenic neuropeptides, normal spontaneous food intake patterns, and growth rates, and a normal hyperphagic response to fasting. These discoveries propose that ghrelin is not imperative in the regulation of appetite.84
Furthermore, differences exist between people in the alteration of subjective desire to eat resulting from ghrelin levels and food restriction. In healthy men, caloric restriction across four days is enough to increase appetite and significantly decrease lean body mass and was not accompanied by alterations to serum ghrelin levels.85 Stronger evidence could be needed to support the suggested role of ghrelin as a ‘hunger signal’ in normal feeding completely.
Many approaches have been employed to block ghrelin activity. GHS-R1a antagonists decrease food intake acutely in lean, diet-induced obese and ob/ob mice, and repeated administration to ob/ob mice leads to reduced weight gain.86 A similar acute result has been exhibited in rats.87 Yet, it seems that not all GHS-R1a antagonists have similar influences on appetite.
For example, BIM-28163, which is a ghrelin analog with full competitive GHS-R1a antagonist characteristics, stops ghrelin-stimulated growth hormone release in rats but stimulates food intake and weight gain.88,89 This suggests the existence of a novel receptor regulating the orexigenic actions of ghrelin.
The utilization of RNA-Spiegelmers (stable oligonucleotides with specific target binding properties) is another method for blocking the orexigenic effects of ghrelin. NOX-B11, a high-affinity Spiegelmer specific for octanoylated ghrelin, reduces ghrelin-induced food intake90 and leads to weight loss in mice with diet-induced obesity.91
Another technique which is under investigation is the utilization of anti-ghrelin ‘vaccines’, which cause weight loss in rats92 and pigs93. A recent phase I/II clinical trial exhibited no evidence of an effect on weight in obese humans, despite displaying a robust antibody response.94
Peptide YY
The endocrine L cells of the large and small bowel produce Peptide YY 3-36 (PYY3-36) in response to the presence of food. It is reported that levels of the peptide increase postprandially and decrease food intake.95
Recently, it has been demonstrated that PYY3-36 is also produced by neurons of the paraventricular nucleus (PVN), ARC, and supraoptic nuclei of the human hypothalamus.96 Based on evolutionary and structural criteria, PYY3-36 is closely related to NPY and pancreatic polypeptide (PP),97 which all act on the NPY receptor family.98
As with leptin, PYY3-36 has been seen to cross the blood-brain barrier and act on the Y2 receptor, a presynaptic inhibitory autoreceptor on NPY neurons.95,99 Activation of Y2 leads to lower NPY release and higher α-MSH release.95 Furthermore, PYY3-36-deficient mice exhibit changes in their energy metabolism, supporting a role for PYY3-36 in the regulation of energy homeostasis.100,101
Low levels of PYY3-36 are seen in obese humans, proposing that a deficiency could contribute to the pathogenesis of obesity. Infusion of PYY3-36 significantly decreases cumulative 24-hour energy intake in both lean and obese subjects. In contrast to the negligible effect on appetite as a result of the daily alterations in circulating leptin, PYY3-36 has been seen to inhibit food intake in humans and rodents at physiological concentrations.
Unlike leptin, in obese subjects, there is no evidence of resistance to PYY3-36.102 It should be noted that central administration of PYY3-36 can stimulate eating, although these results could potentially be of massive importance.103
The absence of obesity-associated resistance to the anorectic properties of PYY3-36 makes it an attractive target for treatment. Currently, intranasal PYY3-36 is the subject of long-term phase II studies. Yet, its employment seems to be hindered by adverse side effects such as nausea and vomiting.104

Pancreatic Polypeptide
Derived from pre-proglucagon, pancreatic polypeptide (PP) is a 36 amino acid peptide. It is released by the pancreatic islet cells in response to food intake and corresponds to the calories ingested. Low levels of PP have been discovered in obese humans and genetically obese mice105 and high levels are present in patients with anorexia nervosa.106
Additionally, peripheral administration of PP has been seen to decrease food intake in obese and lean rodents and ob/ob mice are not as sensitive to the peptide’s actions.107 PP infusion can produce notable, apparently long-lasting intake suppression in humans.108 This has lead to the proposal that PP may behave as a circulating satiety signal.
The mechanism by which PP decreases food intake has not yet been determined, although actions on gastric emptying or regulation of ghrelin, NPY, and orexin have been suggested.109,110 It has been discovered that PP signals via the NPY Y4 and Y5 receptors and so could activate neurons in the hypothalamus directly.111
The suppressive effects of PP have not been consistently replicated, and are relatively modest, even at high doses. The potential role of PP is further complicated by the discovery that the central administration of the peptide can induce moderate hyperphagia, potentially via actions on Y5 receptors.
Although there is some evidence for PP production within the CNS, circulating PP can enter the brain, so clearly there is a requirement for the opposing actions of peripherally and centrally administered exogenous PP to be further examined.
Knowledge of the actions of PP has resulted in the development of two synthetic peptide hormones; TM30338, a dual Y2-Y4 receptor agonist that leads to an acute reduction in food intake, and TM30339, a selective Y4 receptor agonist, which is likely to be the subject of phase I/II study in the near future.104


Glucagon-like Peptide 1
Like PP, glucagon-like peptide 1 (GLP-1) is a derivative of pre-proglucagon, and there are two circulating forms identified in mammals: the predominant GLP-1 (7-36) amide, and GLP-1 (7-37). GLP-1 is co-secreted with PYY3-36 in response to nutrients in the gut, especially carbohydrates.112
Like other gastrointestinal peptides, GLP-1 is also produced in the CNS,113 particularly the nucleus of the solitary tract (NTS) and hypothalamus, with high levels of GLP-1 receptor mRNA present in ARC and PVN. A physiological role of GLP-1 as an anorectic or satiety factor is suggested due to the observations that intracerebroventricular (i.c.v.) injection suppresses food intake and body weight gain in normal and obese rats.
Additionally, daily administration of exendin-(9- 39), a GLP-1 receptor antagonist, augments food intake and body weight.114 The anorectic effects of GLP-1 may be mediated through NPY signaling since GLP-1 inhibits, and exendin-3 (9-39) promotes NPY-induced feeding.115
Exendin-3 (9-39) also blocks leptin-induced inhibition of food intake, and GLP-1 neurons in the NTS co-express leptin receptors, suggesting that the GLP-1 pathway may be one of the mediators of the anorectic effects of leptin.1
Intraventricular GLP-1 powerfully inhibits feeding in rodents, and this response is blocked by the concurrent administration of exendin-3 (9 39). Also, GLP-1 functions as an incretin, enhancing insulin secretion and suppressing glucagon secretion after a meal.116,117 In humans, infusion of GLP-1 at the start of a meal suppresses feelings of hunger and increases satiety scores, without affecting palatability.118
It also causes small dose-dependent inhibition in food intake in both lean and overweight subjects.119 Prandial injections of GLP-1 given to obese but otherwise healthy volunteers for five days resulted in a mean body weight loss of 0.55 kg.120 The combination of enhanced insulin release with a reduction in food intake makes GLP-1 an attractive potential treatment for patients with type II diabetes.
It is important to note some discrepancies that could affect interpretations of the role of GLP-1. For instance, GLP-1 receptor knockout mice do not exhibit any abnormalities in feeding behavior and do not tend to become obese.121
Additionally, GLP-1 induces conditioned taste aversion suggesting that the peptide may suppress feeding by inducing a sensation of sickness.122 Human studies with exendin-4 (exenatide), a naturally occurring peptide with sequence homology to GLP-1, have shown it produces significant reductions in body weight.123 The therapeutic potential of extendin-4 is limited by its side effects, which include nausea and vomiting.124
Liraglutide, another analog of GLP-1, improves glycemic control in association with weight loss. However, similarly to exendin-4, it induces nausea.125 CJC-1134, a newly developed GLP-1 analog, seems to have better tolerability.104
Oxyntomodulin
Oxyntomodulin (OXM) is a 37 amino acid peptide which comes from pre-proglucagon processing in the L cells of the small intestine and in the CNS.126 OXM is released in response to food ingestion and in proportion to meal caloric content.127 Levels are notably elevated in tropical malabsorption and after jejuno-ileal bypass surgery for morbid obesity; which is both conditions associated with weight loss and anorexia.128
Not much is known about its physiological role, despite the high OXM- like immunoreactivity in the CNS, particularly in the hypothalamus. OXM has been shown to cause a sustained and robust inhibition of food intake following central and systemic administration in humans and rats.128-132
In addition, chronic i.c.v. administration results in a notable decrease in body weight gain and adiposity,133 proposing OXM as a possible regulator of body weight and appetite. The anorectic effects of OXM are abolished in GLP-1R(-/-) mice and can be inhibited by exendin-(9-39). This shows that OXM actions are at least partially dependent on GLP-1R, proposing complex interactions of different pre-proglucagon-derived peptides acting at a common target.131
The systemic administration of OXM significantly reduces hunger and food intake in healthy humans.128 The mechanism of action of OXM is still unknown.
It has been proposed that the circulating peptide could access the brain via the ARC and exert its anorectic actions through indirect activation of pro-opiomelanocortin (POMC) neurons in the hypothalamus and through the blocking of fasting ghrelin levels.128 Hence, for the treatment of obesity, OXM could provide a novel route for the development of therapeutic agents.
Amylin
Amylin is an additional pancreatic peptide that decreases food intake. It is a 37 amino acid peptide that belongs to the family of calcitonin gene-related peptides (CGRP) and is a physiological product of pancreatic β cells. It is co-secreted with insulin in a molar ratio that generally stays the same but which may be modified by disease states, including diabetes and obesity.104
Amylin crosses the blood-brain barrier via specific transport systems134 and suppresses feeding in both free-feeding and food-deprived rodents. It is suggested that it acts on receptors in the area postrema (AP),135 although the highest density of amylin binding sites (modified calcitonin receptors, AMY1-3) occur in the hypothalamus. In concordance, amylin-deficient mice exhibit increased weight gain.
Intra-AP treatment with the amylin antagonist AC 187 inhibits the anorectic actions of peripherally administered amylin.136 Crucially, AC 187 heightens food intake when administered alone, either peripherally or centrally, by enlarging meal size and increasing meal frequency.137
Several clinical trials have shown that in diabetic patients, the amylin analog pramlintide leads to a notable decrease in body weight.138 Recently, pramlintide has been granted Food and Drug Administration approval.139

Table 3. Orexigenic peptides. Commonly used doses, agonists, and antagonists.
Cholecystokinin
Cholecystokinin (CCK) is a linear peptide that is synthesized as a pre-prohormone and then cleaved to produce a family of peptides. The predominant forms in plasma are CCK-8, CCK-33, and CCK-39. CCK is generated via endocrine I cell in the jejunum and duodenum and was the first gut hormone which was shown to dose-dependently reduce food intake in several species, including humans.140-142
It has been suggested that it acts as a satiety signal via CCK1 receptor activation on vagal afferents.140 Otsuka Long Evans Tokushima Fatty (OLEFT) rats are insensitive to the anorexigenic action of CCK and lack CCK1 receptors. These animals are obese and hyperphagic, and display deficits in hypothalamic NPY gene expression.143
CCK1 receptor antagonists heighten food intake in several species,144 whereas CCK1 agonists have the opposite effect.145 Peripheral CCK has a fast but relatively short-lived effect on feeding, which is consistent with a role in mediating meal termination and satiety.146 In rats, since reduced meal size is largely compensated for by an increase in meal frequency, CCK administration does not lead to weight reduction.147
In humans, CCK-33 infusion increases feelings of fullness and decreases hunger ratings, while opposite effects have been seen after the infusion of the CCK1 antagonist, loxiglumide.148,149 On the other hand, there is evidence that CCK may play a key part in longer-term energy regulation by synergizing with the actions of leptin.
Central leptin administration potentiates the feeding inhibition of peripheral CCK, and CCK/leptin in combination results in more weight loss over 24 hours than leptin alone. This synergy may happen by CCK activating brainstem neurons which project to the hypothalamus combined with the direct hypothalamic actions of leptin.27

Bombesin and Bombesin Related Peptides
Bombesin is a 14 amino acid amphibian peptide, it has three mammalian analogs: neuromedin B(NMB), neuromedin C (NMC), and gastrin-releasing peptide (GRP). These peptides exert their influence via NMB-preferring bombesin receptor (BB1, NMB- R), the GRP-preferring bombesin receptor (BB2, GRP- R), or the bombesin receptor subtype-3 (bb3, BRS 3).
In several species, including humans, feeding suppression by bombesin/bombesin-like peptides has been recorded.150,151 Peripheral and/or central administration of bombesin/bombesin-like peptides decreases meal size in a dose-dependent manner in rats150 and some other species.152
Infusions of bombesin and GRP decrease food intake in humans by enhancing satiety, although effective doses of bombesin may induce nausea and decrease food palatability.151,153 Certain receptor antagonists can attenuate the anorectic actions of exogenously administered bombesin-like peptides, and the blockade of bombesin receptors within the CNS can cause a huge increase in food intake.
Even though in some cases antagonists for bombesin-like peptide receptors enhance food ingestion, the contribution of endogenous bombesin-like peptides on the normal regulation of food intake is still unclear.152
Examinations utilizing knockout mice may supply new pathways for such research. Deficiencies in BB2and/or bb3 do not influence feeding, although the hypophagic response to low-dose bombesin is suppressed in BB2-deficient mice.154 Though bombesin-like peptides have extremely low affinity for the bb3 receptor, bb3-deficient mice display heightened food consumption and age-related, mild obesity.155
These developments correlate with a heightened hyperphagic response to the orexigen melanin-concentrating hormone (MCH) and levels of hypothalamic MCH receptor and prepro-MCH mRNA are increased.156 Additional studies with bombesin/bombesin-like peptides utilizing both gene-targeting, and traditional pharmacological strategies, could well contribute to the development of new therapeutics for the treatment of obesity.
Conclusion
This article has discussed some of the basic evidence implicating these putative orexigenic and anorexigenic peptides in the complex regulation of body weight, appetite, and energy homeostasis. The list of candidate signals will no doubt expand further, and a variety of non-peptide transmitters exist which have not been discussed, but which are strongly associated with these processes.
It must also be considered that the current emphasis on hypothalamic processes within the brain masks vital influences of extra-hypothalamic circuitry. A better knowledge of the motivational and behavioral aspects of eating control will need a greater understanding of those factors. In relation to several of the peptides discussed here, it is also apparent that the evidence for a primary – or even an actual role – in feeding is sometimes to be questioned.
It is crucial that more thorough analyses of behavior accompany the highly technical assays that link a peptide to a regulatory process largely on the basis of anatomical localization and whether food intake is suppressed or stimulated after exogenous, non-physiological, administration.
Special Mentions
This white paper was based on the original work of:
Sonia A. Tucci, Lynsay Kobelis and Tim C. Kirkham:
Sonia Tucci is a Lecturer in Behavioral Neuroscience and Tim Kirkham is a Professor of Biopsychology, both at the University of Liverpool. Their research interests currently center on the pharmacological analysis of the controls of appetite and ingestive behavior, and particularly the role of central endocannabinoids in the regulation of appetite. Lynsay Kobelis is conducting doctoral research into opioidcannabinoid interactions in feeding.
University of Liverpool, Eleanor Rathbone Building, Bedford Street South, UK

About Tocris Bioscience
Tocris Bioscience is your trusted supplier of high-performance life science reagents, including receptor agonists & antagonists, enzyme inhibitors, ion channel modulators, fluorescent probes & dyes, and compound libraries. Our catalog consists of over 4,500 research tools, covering over 400 protein targets enabling you to investigate and modulate the activity of numerous signaling pathways and physiological processes.
We have been working with scientists for over 30 years to provide the life science community with research standards, as well as novel and innovative research tools. We understand the need for researchers to trust their research reagents, which is why we are committed to supplying our customers with the highest quality products available, so you can publish with confidence.
Tocris is part of the protein sciences division of Bio-Techne, which also includes the best in class brands R&D Systems, Novus Biologicals, ProteinSimple, and Advanced Cell Diagnostics. Bio-Techne has united these brands to provide researchers with a full portfolio of research reagents, assays, and protein platforms. For more information on Bio-Techne and its brands, please visit bio-techne.com.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices, and treatments.