Cell and Gene Therapy (CGT) emerged as a potential treatment for blood-based cancers via the FDA’s accelerated approval process. However, CGT was quickly reined in even as it heralded a wave of next-generation biologic-based treatments. Control measures were quickly brought in due to the ever-shifting landscape of regulatory guidelines that ensure patient safety.
Regulatory agencies in the United States and Europe have published guidelines established to help regulate various areas of this rapidly expanding field. Within this complex regulatory framework, securing Investigational New Drug (IND) approval for CGT products is a challenging task.
When securing IND approval, CGT manufacturers tend to overlook the quality control measures used against manufacturing the raw materials they have sourced. Regarding Good Manufacturing Practice (GMP) products, ACRO Biosystems can guarantee they have conducted extensive research to meet the standards set by global regulations related to raw or ancillary materials.
All raw materials have been designed according to regulatory requirements from product inception to commercialization. Therefore, ACRO Biosystems’s ancillary materials are among the highest GMP standards in line with the mandates of the various global regulatory bodies.
This article will outline which regulatory guidelines were followed for manufacturing ACRO Biosystems’s GMP-grade products.
Compliance requirements for GMP ancillary materials in CGT manufacturing
GMP-grade products across the industry should meet the recommendations of the pharmacopeia documents, which govern various vital aspects of raw materials, as shown in the table below:
1. USP<1043>ancillary materials for cell, gene and tissue-engineered products
Source: ACROBiosystems

Ancillary materials (AM) are placed into four categories in accordance with USP 1043 based on the significance of their role in Cell and Gene Therapy (CGT) processes and risk assessment.
Important documentation, such as Drug Master Files (DMF), Certificates of Analysis (CoA), and guarantees that AM with animal or human-derived materials has been purified, tested, and is free of external exogenous factors, are emphasized.
Most cytokines for cell culture additives are classified as Tier 2 products, where regulations play a crucial role in ensuring the traceability, quality, and safety of ancillary materials, highlighting transparency and reliability in CGT manufacturing processes.
2. IPRP (International Pharmaceutical Regulators Programme) “general considerations for raw materials used in the production of human cell and gene therapy products”
• Establishing a quality management system
The IPRP report, in Section 3.1, calls for CGT manufacturers to implement a strict quality management system, covering all raw material certification programs. This contains basic, key facts such as supplier approval, execution of quality agreements, provision of CoA, and supply of necessary supporting documents.
As such, the CGT manufacturer bears the responsibility to carry out all due diligence when it comes to identifying an appropriate raw materials supplier. This means evaluating how suppliers and manufacturers comply with raw material storage requirements, stability control, and the management of expiration dates.
• Ensuring transparency of origin and traceability
Section 3.3 outlines the importance of origin and traceability considerations. Raw materials should always be supported with the right documents, including Certificates of Origin (COO), which facilitate traceability and identification material or component origin used in the production process.
When materials from human or animal sources are used, it is recommended that CGT product manufacturers selectively opt for materials with all source information supplied. This may involve agreements with suppliers, such as ensuring certain materials are sourced from specific regions to ensure a transparent and traceable supply chain.
• Microbial safety in raw material production
The report (Section 4.2) highlights a significant issue when addressing microbial safety concerns during raw material production: many CGT products do not experience terminal sterilization. Therefore, implementing robust, sterile testing methods for raw materials used in CGT product production is vital.
Given the limitations of conventional virus inactivation/removal procedures for CGT products, the report calls for verifying methods for virus inactivation/removal steps when using raw materials that have been biologically sourced.
In cases where such procedures are unworkable, and in addition to establishing a justified rationale for their usage, thorough testing for the presence of relevant viruses in raw materials should still be performed. This multipronged stringent approach ensures the safety of raw materials crucial to producing advanced Cell and Gene Therapy products.
3. European pharmacopeia 5.2.12 “raw materials for the production of cell-based and gene therapy medicinal products”
The pharmacopeia document highlights the stringent requirements applied across the EU region related to raw materials of biological origin used in the production of CGT products across their lifecycle.
Section 3.2 specifically addressing production, and major considerations include the need for production within a suitable quality management system and facility. This should allow for the integration of a rigorous aseptic production process that takes into account how additives, such as antibiotics, influence CGT products.
General quality requirements for raw materials cover a large set of criteria, including biological activity (specific activity,content, identification, impurities (including residual host cell protein/DNA), microorganisms, mycoplasmas, purity, virus contaminants, and water purity. All in all, screening for this criteria ensures the integrity and safety of raw materials throughout the entire production process.
A major emphasis anchors itself on the sterility of raw materials, mandating for the production of materials as sterile or terminally sterilized under aseptic conditions unless clearly stated otherwise.
If a case arises where a raw material is not classed as sterile, a prerequisite is the knowledge and disclosure of the level of microbial contamination. This comprehensive approach to raw material specifications neatly aligns with the primary objective; ensuring that the CGT manufacturing process meets the highest standards in quality and safety from beginning to end.
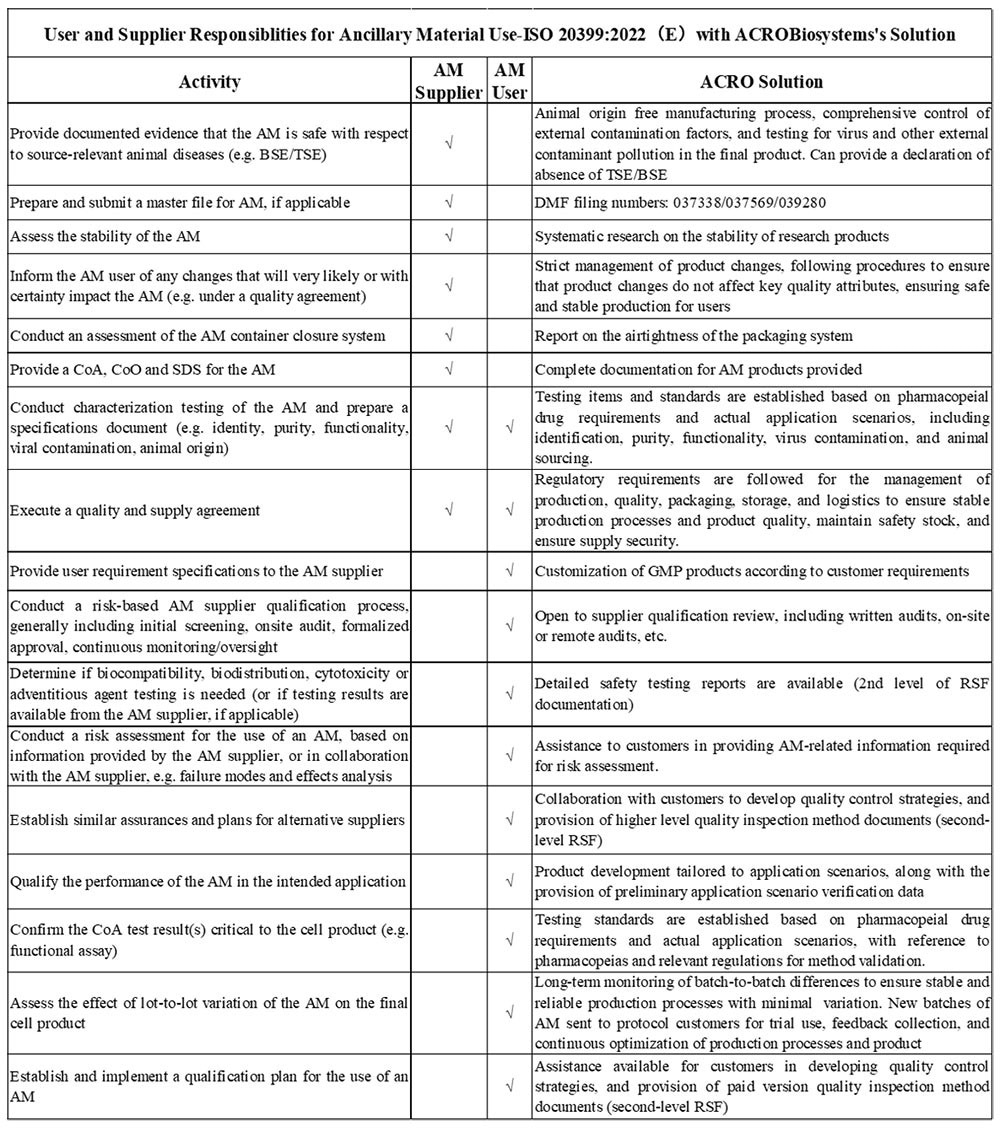
4. ISO 20399:2022 (E) “Biotechnology - Ancillary materials present during the production of cellular therapeutic products and gene therapy products”
The International Organization for Standardization introduced ISO 20399:2022(E), offers detailed framework that outlines the roles of AM suppliers and users in the manufacture of CGT products.

Image Credit: ACROBiosystems
Due to the complexities involved in the interchange between supplier and manufacturer, ACRO Biosystems is dedicated to refining this process. With tailored solutions and support, ACRO Biosystems is on hand to carefully navigate its most valued customers through these requirements. The company is committed to keeping up-to-date with industry regulations to provide the appropriate support and help its clients deliver a smooth and compliant production process.
ACRO Biosystems has taken the time to evaluate the most important guidelines established by regulatory bodies, to help its customers meet regulatory filing requirements in both the United States and the EU.
Here are a few ways which highlight why ACRO Biosystems is a valuable partner in navigating the regulatory landscape for CGT production.
a. Comprehensive regulatory support documents:
The cornerstone of ACRO Biosystems’ capabilities is the conscientious gathering of Regulatory Support Files (RSF), which corresponds with international regulations and official requirements while meeting a customer’s needs. These documents carefully detail the essential qualifications required for GMP-grade materials in CGT production, tailored to meet the varied requirements of customers across a diversity of developmental stages.
Level 1 documents:
Level One documents cover product qualifications. These are supplied free of charge to all customers who have bought GMP products. Across 40-60 pages, these documents have been developed to address the needs of customers in the research and pre-clinical stages. They essentially act as proof of supplier qualifications throughout the material screening phase.

Image Credit: ACROBiosystems
Level 2 documents:
Level Two documents are unique quality and safety documents. Comprised of more than 1000 pages, these documents can be purchased by customers entering the clinical phase. At this stage, customers need comprehensive knowledge on raw materials to support their clinical applications or market authorization.
Level Two documents include detailed analytical method validation reports and Standard Operating Procedures (SOPs), as well as allowing customers to save time, costs, and labor in material analysis development and validation procedures.

Image Credit: ACROBiosystems
b. Comprehensive GMP products with U.S. FDA filing support:
ACRO Biosystems actively supports Drug Master File (DMF) filing, a complete set of documents that detail the information on product chemistry, manufacturing, and controls (CMC) information. ACRO Biosystems is the first company to secure FDA DMF filing for recombinant protein reagents, which it implements across all GMP products and other recombinant proteins.
Source: ACROBiosystems
c. IND/new drug application (NDA)/biological license application (BLA) stage compliance support:
ACRO Biosystems offers on-site support to help customers with the IND/NDA/BLA filing process. This service includes addressing regulatory queries related to ancillary materials, so all ACRO Biosystems customers can ensure they have the relevant documentation needed for their filings.
Summary
This article comprehensively outlines the complex regulatory landscape of Cell and Gene Therapy (CGT) manufacturing, with a key focus on meeting compliance within the stringent frameworks of regulatory bodies in both the US and EU.
Carving out a smooth path on the way to IND approval comes from understanding the supplier-manufacturing relationship and the appropriate checks and balances behind manufacturing CGT products.
This evaluation of key regulatory guidelines, which covers USP, IPRP, European Pharmacopeia, and ISO, illustrates the criteria that govern ancillary materials while clarifying the steps that must be taken to navigate the regulatory landscapes of CGT production. Resultingly, ACRO Biosystems can help cater to the diverse needs of its customers across all developmental stages of the CGT production process.
About ACROBiosystems
ACROBiosystems is a cornerstone enterprise of the pharmaceutical and biotechnology industries. Their mission is to help overcome challenges with innovative tools and solutions from discovery to the clinic. They supply life science tools designed to be used in discovery research and scalable to the clinical phase and beyond. By consistently adapting to new regulatory challenges and guidelines, ACROBiosystems delivers solutions, whether it comes through recombinant proteins, antibodies, assay kits, GMP-grade reagents, or custom services. ACROBiosystems empower scientists and engineers dedicated towards innovation to simplify and accelerate the development of new, better, and more affordable medicine.
Sponsored Content Policy: News-Medical.net publishes articles and related content that may be derived from sources where we have existing commercial relationships, provided such content adds value to the core editorial ethos of News-Medical.Net which is to educate and inform site visitors interested in medical research, science, medical devices and treatments.